CME #135 June 2008
Skin & Aging is proud to bring you this latest installment in its CME series. This series consists of regular CME activities that qualify you for 1 AMA PRA Category 1 Credit. As a reader of Skin & Aging, this course is brought to you free of charge — you aren’t required to pay a processing fee. In this CME, Drs. Sarah Walsh and Omar Sangüeza discuss nephrogenic systemic fibrosis (NSF), an emerging fibrosing condition that primarily affects the skin and has recently been linked to exposure to gadolinium-containing contrast agents. At the end of this article, you’ll find an exam. Mark your responses in the designated area, then fax page 41 to NACCME at (610) 560-0501.
We’ll also post this course on our Web site, which you can access at www.skinandaging.com. I hope this CME contributes to your clinical skills.
Amy McMichael, M.D. CME Editor
Amy McMichael, M.D., is Associate Professor in the Department of Dermatology, Director of the Hair Disorders Clinic and Residency Program Director at Wake Forest University Medical Center in Winston-Salem, NC.
Nephrogenic Systemic Fibrosis: A Comprehensive Review
By Sarah N. Walsh, M.D., and Omar P. Sangüeza, M.D.
Nephrogenic systemic fibrosis (NSF), initially referred to as nephrogenic fibrosing dermopathy (NFD), is an emerging fibrosing condition that primarily affects the skin and occurs in patients with renal injury. Recently, this disease has been linked to exposure to gadolinium-containing contrast agents. The disease can have a rapid or slowly progressive course, which generally begins with pain, pruritus, and swelling of the extremities and trunk and evolves to hardened woody induration with joint contractures, with or without systemic involvement. While partial or complete spontaneous remission has been reported, numerous other therapies have been tried with varying degrees of success. To date, prevention appears to be the most effective option. Significant morbidity and an increased mortality are the most pressing concerns, leading to continuous active research to further identify risk factors, inciting agents, pathogenesis, and effective treatment modalities.
Historical Perspective
In a Research Letter to the Lancet in 2000, Cowper et al reported on 15 renal dialysis patients who had a new cutaneous fibrosing disorder.1 It was recognized to be similar to scleromyxedema; however, the main distinguishing features were: 1. absence of a serum monoclonal paraprotein; 2. lack of systemic mucin deposition; and 3. sparing of the face. Shortly thereafter, it was again Cowper et al who wrote the seminal article “Nephrogenic Fibrosing Dermopathy,” which put a name to this new entity.2 This 2001 paper described indurated plaques and papules that developed on the trunk and extremities of patients with renal dysfunction. Histologically, the biopsies showed thickened dermal collagen with widening of subcutaneous septa and increased dermal mucin and elastic fibers. Stellate spindled cells that were CD34 positive were intermingled among the large dermal collagen bundles. Only one case in the series showed spontaneous resolution after dialysis for acute tubular necrosis.
In 2003, an explosion of published articles contributed to a wider understanding of NFD. In July, Ting et al reported on the first autopsy of a patient with NFD.3 This was significant as it provided the first undisputed evidence of systemic involvement in which extensive fibrosis and calcification were noted in the diaphragm, psoas muscle, renal tubules, and rete testes.
A Letter to the Editor by Cowper and Bucala in the American Journal of Dermatopathology in August hypothesized on the culprit cell of NFD.4 By using a dual immunohistochemical staining technique with CD34 and procollagen, they found that the majority of the dermal spindle cells were positive for both markers. These cells were thereby identified as circulating fibrocytes, which were first described in the rheumatology literature in 1994 and known to be bone marrow-derived cells that circulate as fibroblast progenitor cells, produce collagen, and aid in wound repair and remodeling.5
Since the majority of the reported cases to this point had occurred in patients receiving dialysis for severe renal disease, in 2004 Jimenez et al proposed a new terminology for the disorder, dialysis-associated systemic fibrosis (DASF).6 However, concurrent and subsequent reports demonstrated that this disease affects patients with a wide spectrum of acute and chronic renal disorders that did not necessarily require dialysis. In addition, further evidence supported systemic involvement. Therefore in 2005, the name of the disease was more accurately changed to nephrogenic systemic fibrosis.7
Gadolinium is a naturally occurring earth element that is a part of the lanthanoid series (atomic #64).9 This silvery white metal has seven unpaired electrons, which make it highly paramagnetic and therefore ideal to enhance images in magnetic resonance (MR) scanning. In 1988, gadolinium-based contrast agents were first used in humans.8 The use of these agents increased, and in 1997, the first paper implementing their utility in renal patients was published.10
While Cowper et al speculated that a “recently introduced material, possibly a contrast agent . . .” stimulated circulating fibrocytes, culminating in the fibrotic changes, it was Grobner et al who, in 2006, provided evidence of the first real connection between gadolinium and NSF.11,12 This connection was solidified with subsequent studies in which gadolinium was detected within tissues of patients with NSF.13
As evidence for this association accumulated, the U.S. Food and Drug Administration responded by issuing a public health advisory warning on the use of gadolinium-based contrast agents in patients with renal dysfunction.14
The 2007 guidelines of the European Society of Urogenital Radiology stated that “gadodiamide [the generic name for Omniscan] should not be administered to patients with reduced kidney function or to those on dialysis.”15
In addition to the discovery of the link between gadolinium and NSF in 2006, this year was also significant for publications that documented cases in other parts of the world — including Europe and India — as well as identification of the disease in children.16,17
The most recent reports have focused on other associations or risk factors for the development of NSF as well as effective treatment options.
Epidemiology
Yale researchers have maintained a registry called the International Center for Nephrogenic Fibrosing Dermopathy Research, which has identified more than 215 cases thus far (last updated Jan. 20, 2008).18 This condition seems to occur in approximately 4% of patients with advanced or end-stage renal disease.19 Patients have ranged in age from 8 to 86 years.20 Males and females are equally affected (M:F ratio of 1:1), and no racial predilection has been noted.20
Pathogenesis
The circulating fibrocyte has been implicated in the development of NSF. These cells have features of both connective tissue cells (procollagen-positive) and bone marrow derived progenitor cells (CD34-positive), hence the name fibrocyte, which combines fibroblast with leukocyte/thrombocyte/erythrocyte.21
Circulating fibrocytes produce chemokines and growth factors, including transforming growth factor-beta (TGF-β), which induces connective tissue matrix production as well as angiogenesis. Since these cells are not noted to be mitotically active on microscopic examination, it is speculated that they are recruited from the circulation, deposit collagen, and eventually mature into fibroblasts or myofibroblasts, at which point they lose their CD34 phenotype.21
The link between NSF and gadolinium-based contrast agents is now well established. Clearance of gadolinium is significantly reduced in the setting of renal dysfunction. Gadolinium has been detected in the insoluble unchelated toxic form (Gd3+) by quantitative scanning electron microscopy/energy-dispersive X-ray spectroscopy in the tissues of patients with NSF.22 In addition, higher dermal concentrations of gadolinium have been found in late lesions when compared with biopsies taken at the onset of the disease; this is thought to be from initial bone storage with subsequent mobilization.22 This free (unchelated) toxic Gd3+ is postulated to act as a target or trigger for recruitment of circulating fibrocytes.12
A recent article by Parsons et al has suggested another possible trigger in the pathogenesis of NSF.23 Through immunohistochemical staining methods, these authors found an increased expression of transglutaminases in biopsy specimens with the diagnosis of NSF. Gadolinium has been shown to activate transglutaminases, and transglutaminases in turn are known to activate TGF-β.24,25 As discussed previously, TGF-β induces fibroblastic proliferation.
While the exact stepwise development of the disease is still under investigation, renal dysfunction, gadolinium, circulating fibrocytes, TGF-β, and transglutaminases all seem to play a role, either in a dominant fashion or synergistically.
Risk Factors
Renal dysfunction is a prerequisite for NSF. It was once thought that only patients with severe renal disease — particularly those requiring dialysis — were affected. Subsequently, it became clear that any duration or degree of renal injury was sufficient. However, it has been well established that those at greatest risk are patients with chronic kidney disease or end-stage renal disease (ESRD) — particularly those with a glomerular filtration rate of <30 ml/min/1.73 m2 as well as those receiving peritoneal dialysis (even more so than hemodialysis).19
As highlighted previously, exposure to gadolinium is another monumental risk factor. While there have been reported cases of NSF occurring in patients without known exposure to the element, the vast majority have had at least one scan or procedure in which a gadolinium-based contrast agent was injected.26 The absolute risk for developing NSF in patients with ESRD has been reported as 2.4% per single MR study.27
The type of gadolinium-containing contrast agent used is also important. There are currently five agents approved by the FDA for use in humans. These differ mainly in relation to transmetallation and kinetic and thermodynamic stability (amount of free toxic Gd3+ released from the chelate by exchange with another metal).9 Gadodiamide (Omniscan) has a low thermodynamic stability and demonstrates more transmetallation, accounting for this agent being associated with the largest number of reported cases of NSF.9
There are additional factors to consider in relation to the use of gadolinium. Collidge et al found a positive association between the cumulative dose of gadolinium and the development of NSF.28 In patients with severe liver disease, the glomerular filtration rate is often overestimated, resulting in a greater risk of gadolinium exposure in this subgroup.29 Children less than 1 year old are also at a higher risk due to immature renal function.30
A number of other factors have been discussed as potentially contributing to the development of NSF. These include coexistent acidosis, erythropoietic stimulating agents (recombinant human erythropoietin), major proinflammatory events (major surgery, infection/sepsis, vascular events, or thrombosis), a hypercoagulable state, hyperphosphatemia, medications that could cause transmetallation of gadolinium (iron, zinc, copper, calcium, lanthanum, and sevelamer) and concomitant immunosuppression.31-33 While such conditions and circumstances have been noted in various reports, these have not been found to be consistently reproducible.
Clinical Features
The clinical findings in NSF can be quite inconspicuous in the early stages, yet they may progress to pronounced cutaneous manifestations in the late phases. Initially, patients typically complain of pruritus and/or pain, which have been described as numbness, tingling, burning, sharp, and sizzling.34 In well-developed NSF, patients can have joint contractures and report loss in range of motion with limited mobility.
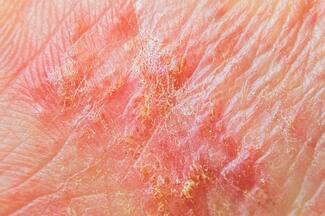
On physical examination, the predominant sites of involvement are the bilateral lower and upper extremities. The trunk can be affected as well; however, the face is characteristically spared. Early lesions may present as subtle superficial papules and plaques and/or focal areas of hair loss (Figure 1). Scattered depigmented scar-like macules can also be seen (Figure 2). Edema of the lower extremities is commonly noted, which is increased over that found at baseline for many of the patients with chronic renal failure. Lesions of longer duration show deep dermal and subcutaneous induration, resulting in a hardened woody consistency (Figures 3a and 3b). Girardi recently described a phenomenon noted in his NSF patients that he termed “banding” whereby the underlying fibrosis pulls the skin down in a linear band.34 The overlying skin can have irregular erythema or velvety discoloration and often becomes dimpled, imparting a cobblestone appearance likened to the skin of an orange (peau d’ orange) (Figure 4). In the most severe forms, patients develop severe disabling joint contractures.
Ocular lesions have been reported, which take the form of whitish-yellow scleral plaques with vascular telangiectasias.35 Visceral involvement is variable; however, fibrosis has been documented in the heart, lungs, liver, kidneys, testes, and skeletal muscles.3,36,37
Histopathological Features
Microscopic examination of cutaneous lesional biopsies shows dermal and subcutaneous fibrosis. The thickened collagen bundles primarily occupy the mid and lower dermis with widening of the subcutaneous septa (Figure 5). Occasionally, there is extension into the underlying fascia or skeletal muscle. An increased deposition of dermal mucin and elastic fibers is seen. In addition, there is a proliferation of histiocytes and dermal dendrocytes, including multinucleated forms. The characteristic finding, however, is the presence of bland non-mitotically active spindle cells scattered within the fibrotic background (Figure 6).
Special and immunohistochemical stains can assist in highlighting these features. Stains for mucin, such as colloidal iron, will confirm the increase in dermal mucin (Figure 7). An elastic stain, such as Verhoff-van Gieson, can demonstrate thickened and more pronounced elastic tissue fibers within the dermis. A CD34 immunohistochemical stain will decorate the dermal spindle cell proliferation. However, if confirmation of these cells as circulating fibrocytes is sought, dual immunohistochemical staining with CD34 and procollagen will be positive. The augmentation of histiocytes and dermal dendrocytes can be established by positive staining with CD68 and Factor XIIIa.
Differential Diagnosis
The differential diagnosis of similar fibrosing cutaneous conditions mainly includes scleromyxedema, systemic sclerosis/morphea, eosinophilic fasciitis, and lipodermatosclerosis.
Scleromyxedema. Histologically, differentiating NSF from scleromyxedema can be the most challenging. A 2005 paper by Kucher et al focused on just that.38 The authors concluded that there were no pathognomonic microscopic features to reliably distinguish between the two entities, either on hematoxylin and eosin examination or by immunohistochemical staining profiles.
Therefore, the mainstay and most accurate way to separate NSF from scleromyxedema are by clinical history and findings. Patients with scleromyxedema have systemic manifestations, most notably a serum monoclonal paraprotein as well as prominent mucin deposition in numerous visceral organs. The cutaneous lesions have a predilection for the arms, hands, and trunk and classically involve the face, which has been universally spared thus far in all reported cases of NSF.
Systemic sclerosis/morphea is an autoimmune connective tissue disorder with similar clinical manifestations of symmetrical hardening of the skin. In addition to cutaneous involvement, the systemic disease (systemic sclerosis) affects the blood vessels and visceral organs, including the gastrointestinal tract, heart and kidneys.
Positive serum markers include antinuclear antibody, anticentromere antibody, and Scl-70. The localized form (morphea) is limited to the skin and presents with indurated plaques on the trunk and extremities. The histology differs in that there is a decrease in dermal spindle cells and elastic fibers as well as the consistent presence of plasma cells.
Eosinophilic fasciitis is another entity resulting in hardened woody induration of the skin with systemic evidence. First described by Shulman in 1974, this syndrome is characterized by fasciitis and fibrosis of the extremities (but with sparing of the hands and feet), polyclonal hypergammaglobulinemia, and peripheral eosinophilia.39
Under the microscope, the thickened collagen bundles layer through the subcutaneous tissue in a roughly parallel fashion with involvement of the underlying fascia and subfascial muscle. Conspicuous eosinophils are typically noted within the accompanying inflammatory infiltrate. Increased dermal mucin is not a feature.
Lipodermatosclerosis, also known as sclerosing panniculitis, is a chronic inflammatory and fibrosing condition of the bilateral lower extremities, classically occurring in obese adult females with a history of venous insufficiency.
The clinical appearance is of erythematous indurated painful woody plaques confined to the distal lower extremities.
The histology is quite dissimilar from that seen in NSF and is typified by exclusive involvement of lobular and septal subcutaneous tissue with fibrosis, fat necrosis, macro- and microcyst formation and lipomembranous change.
Treatment
Numerous forms of therapy have been tried with variable degrees of success. Partial or complete spontaneous remission has occurred, especially with resolution of the underlying renal impairment. Treatments range from supportive measures with physical therapy and pain management to more aggressive regimens, including extracorporeal photophoresis, high-dose intravenous immunoglobulin, immunostimulants (interferon-α), immunosuppressants (cyclosporine, methotrexate, mycofenolate mofetil, and oral and topical corticosteroids), pentoxifylline, thalidomide, photodynamic therapy, psoralen combined with UVA therapy, intravenous sodium thiosulfate, and ACE- inhibitors.12,18,19,26,40-41
Prevention
The most important and effective modality is prevention. Much of the recent research and literature has been dedicated to this crucial aspect. Some of the suggestions have been use of less toxic or implicated gadolinium-containing contrast agents, switch to alternative imaging procedures that avoid or limit exposure to gadolinium, giving the lowest possible dose and avoiding repeat administration when gadolinium-containing contrast agents are absolutely necessary, initiating hemodialysis after gadolinium-requiring imaging procedures to expedite rapid clearance, optimizing patient metabolic panels and treating underlying inflammatory or thrombotic conditions prior to injection of gadolinium, and avoidance of drugs that increase transglutaminases or TGF-β or that could be involved in transmetallation.15,19,23,32,42
Prognosis
The fibrosis in NSF can be rapidly progressive or quiescent and stable. Spontaneous remission has been known to occur. However, there is significant morbidity associated with well-developed cases, mainly as a result of loss in range of motion and mobility that accompanies disabling joint contractures. Systemic involvement can cause fibrosis in numerous visceral organs, leading to severe impairment and dire consequences. An increased mortality rate is associated with the disease, ranging from 7.7% to 67%.19
Conclusion
Nephrogenic systemic fibrosis is a debilitating disease occurring in patients with renal dysfunction that is relatively new to the medical community. Cutaneous involvement is inevitably present, with systemic complications being less frequent. While the details regarding the risk factors, pathogenesis, and effective treatment are still being explored, awareness and recognition of this entity is important in order to take the appropriate measures to prevent disease initiation or progression.
Dr. Walsh is a Dermatopathology Fellow, Departments of Dermatology and Pathology, Wake Forest University School of Medicine, Winston-Salem, NC.
Dr. Sangüeza is Professor, Departments of Dermatology and Pathology and Director of Dermatopathology, Wake Forest University School of Medicine, Winston-Salem, NC.
DISCLOSURE: Dr. Walsh and Dr. Sangüeza have no conflict of interest with any subjects discussed in this article.
CME #135 June 2008
Skin & Aging is proud to bring you this latest installment in its CME series. This series consists of regular CME activities that qualify you for 1 AMA PRA Category 1 Credit. As a reader of Skin & Aging, this course is brought to you free of charge — you aren’t required to pay a processing fee. In this CME, Drs. Sarah Walsh and Omar Sangüeza discuss nephrogenic systemic fibrosis (NSF), an emerging fibrosing condition that primarily affects the skin and has recently been linked to exposure to gadolinium-containing contrast agents. At the end of this article, you’ll find an exam. Mark your responses in the designated area, then fax page 41 to NACCME at (610) 560-0501.
We’ll also post this course on our Web site, which you can access at www.skinandaging.com. I hope this CME contributes to your clinical skills.
Amy McMichael, M.D. CME Editor
Amy McMichael, M.D., is Associate Professor in the Department of Dermatology, Director of the Hair Disorders Clinic and Residency Program Director at Wake Forest University Medical Center in Winston-Salem, NC.
Nephrogenic Systemic Fibrosis: A Comprehensive Review
By Sarah N. Walsh, M.D., and Omar P. Sangüeza, M.D.
Nephrogenic systemic fibrosis (NSF), initially referred to as nephrogenic fibrosing dermopathy (NFD), is an emerging fibrosing condition that primarily affects the skin and occurs in patients with renal injury. Recently, this disease has been linked to exposure to gadolinium-containing contrast agents. The disease can have a rapid or slowly progressive course, which generally begins with pain, pruritus, and swelling of the extremities and trunk and evolves to hardened woody induration with joint contractures, with or without systemic involvement. While partial or complete spontaneous remission has been reported, numerous other therapies have been tried with varying degrees of success. To date, prevention appears to be the most effective option. Significant morbidity and an increased mortality are the most pressing concerns, leading to continuous active research to further identify risk factors, inciting agents, pathogenesis, and effective treatment modalities.
Historical Perspective
In a Research Letter to the Lancet in 2000, Cowper et al reported on 15 renal dialysis patients who had a new cutaneous fibrosing disorder.1 It was recognized to be similar to scleromyxedema; however, the main distinguishing features were: 1. absence of a serum monoclonal paraprotein; 2. lack of systemic mucin deposition; and 3. sparing of the face. Shortly thereafter, it was again Cowper et al who wrote the seminal article “Nephrogenic Fibrosing Dermopathy,” which put a name to this new entity.2 This 2001 paper described indurated plaques and papules that developed on the trunk and extremities of patients with renal dysfunction. Histologically, the biopsies showed thickened dermal collagen with widening of subcutaneous septa and increased dermal mucin and elastic fibers. Stellate spindled cells that were CD34 positive were intermingled among the large dermal collagen bundles. Only one case in the series showed spontaneous resolution after dialysis for acute tubular necrosis.
In 2003, an explosion of published articles contributed to a wider understanding of NFD. In July, Ting et al reported on the first autopsy of a patient with NFD.3 This was significant as it provided the first undisputed evidence of systemic involvement in which extensive fibrosis and calcification were noted in the diaphragm, psoas muscle, renal tubules, and rete testes.
A Letter to the Editor by Cowper and Bucala in the American Journal of Dermatopathology in August hypothesized on the culprit cell of NFD.4 By using a dual immunohistochemical staining technique with CD34 and procollagen, they found that the majority of the dermal spindle cells were positive for both markers. These cells were thereby identified as circulating fibrocytes, which were first described in the rheumatology literature in 1994 and known to be bone marrow-derived cells that circulate as fibroblast progenitor cells, produce collagen, and aid in wound repair and remodeling.5
Since the majority of the reported cases to this point had occurred in patients receiving dialysis for severe renal disease, in 2004 Jimenez et al proposed a new terminology for the disorder, dialysis-associated systemic fibrosis (DASF).6 However, concurrent and subsequent reports demonstrated that this disease affects patients with a wide spectrum of acute and chronic renal disorders that did not necessarily require dialysis. In addition, further evidence supported systemic involvement. Therefore in 2005, the name of the disease was more accurately changed to nephrogenic systemic fibrosis.7
Gadolinium is a naturally occurring earth element that is a part of the lanthanoid series (atomic #64).9 This silvery white metal has seven unpaired electrons, which make it highly paramagnetic and therefore ideal to enhance images in magnetic resonance (MR) scanning. In 1988, gadolinium-based contrast agents were first used in humans.8 The use of these agents increased, and in 1997, the first paper implementing their utility in renal patients was published.10
While Cowper et al speculated that a “recently introduced material, possibly a contrast agent . . .” stimulated circulating fibrocytes, culminating in the fibrotic changes, it was Grobner et al who, in 2006, provided evidence of the first real connection between gadolinium and NSF.11,12 This connection was solidified with subsequent studies in which gadolinium was detected within tissues of patients with NSF.13
As evidence for this association accumulated, the U.S. Food and Drug Administration responded by issuing a public health advisory warning on the use of gadolinium-based contrast agents in patients with renal dysfunction.14
The 2007 guidelines of the European Society of Urogenital Radiology stated that “gadodiamide [the generic name for Omniscan] should not be administered to patients with reduced kidney function or to those on dialysis.”15
In addition to the discovery of the link between gadolinium and NSF in 2006, this year was also significant for publications that documented cases in other parts of the world — including Europe and India — as well as identification of the disease in children.16,17
The most recent reports have focused on other associations or risk factors for the development of NSF as well as effective treatment options.
Epidemiology
Yale researchers have maintained a registry called the International Center for Nephrogenic Fibrosing Dermopathy Research, which has identified more than 215 cases thus far (last updated Jan. 20, 2008).18 This condition seems to occur in approximately 4% of patients with advanced or end-stage renal disease.19 Patients have ranged in age from 8 to 86 years.20 Males and females are equally affected (M:F ratio of 1:1), and no racial predilection has been noted.20
Pathogenesis
The circulating fibrocyte has been implicated in the development of NSF. These cells have features of both connective tissue cells (procollagen-positive) and bone marrow derived progenitor cells (CD34-positive), hence the name fibrocyte, which combines fibroblast with leukocyte/thrombocyte/erythrocyte.21
Circulating fibrocytes produce chemokines and growth factors, including transforming growth factor-beta (TGF-β), which induces connective tissue matrix production as well as angiogenesis. Since these cells are not noted to be mitotically active on microscopic examination, it is speculated that they are recruited from the circulation, deposit collagen, and eventually mature into fibroblasts or myofibroblasts, at which point they lose their CD34 phenotype.21
The link between NSF and gadolinium-based contrast agents is now well established. Clearance of gadolinium is significantly reduced in the setting of renal dysfunction. Gadolinium has been detected in the insoluble unchelated toxic form (Gd3+) by quantitative scanning electron microscopy/energy-dispersive X-ray spectroscopy in the tissues of patients with NSF.22 In addition, higher dermal concentrations of gadolinium have been found in late lesions when compared with biopsies taken at the onset of the disease; this is thought to be from initial bone storage with subsequent mobilization.22 This free (unchelated) toxic Gd3+ is postulated to act as a target or trigger for recruitment of circulating fibrocytes.12
A recent article by Parsons et al has suggested another possible trigger in the pathogenesis of NSF.23 Through immunohistochemical staining methods, these authors found an increased expression of transglutaminases in biopsy specimens with the diagnosis of NSF. Gadolinium has been shown to activate transglutaminases, and transglutaminases in turn are known to activate TGF-β.24,25 As discussed previously, TGF-β induces fibroblastic proliferation.
While the exact stepwise development of the disease is still under investigation, renal dysfunction, gadolinium, circulating fibrocytes, TGF-β, and transglutaminases all seem to play a role, either in a dominant fashion or synergistically.
Risk Factors
Renal dysfunction is a prerequisite for NSF. It was once thought that only patients with severe renal disease — particularly those requiring dialysis — were affected. Subsequently, it became clear that any duration or degree of renal injury was sufficient. However, it has been well established that those at greatest risk are patients with chronic kidney disease or end-stage renal disease (ESRD) — particularly those with a glomerular filtration rate of <30 ml/min/1.73 m2 as well as those receiving peritoneal dialysis (even more so than hemodialysis).19
As highlighted previously, exposure to gadolinium is another monumental risk factor. While there have been reported cases of NSF occurring in patients without known exposure to the element, the vast majority have had at least one scan or procedure in which a gadolinium-based contrast agent was injected.26 The absolute risk for developing NSF in patients with ESRD has been reported as 2.4% per single MR study.27
The type of gadolinium-containing contrast agent used is also important. There are currently five agents approved by the FDA for use in humans. These differ mainly in relation to transmetallation and kinetic and thermodynamic stability (amount of free toxic Gd3+ released from the chelate by exchange with another metal).9 Gadodiamide (Omniscan) has a low thermodynamic stability and demonstrates more transmetallation, accounting for this agent being associated with the largest number of reported cases of NSF.9
There are additional factors to consider in relation to the use of gadolinium. Collidge et al found a positive association between the cumulative dose of gadolinium and the development of NSF.28 In patients with severe liver disease, the glomerular filtration rate is often overestimated, resulting in a greater risk of gadolinium exposure in this subgroup.29 Children less than 1 year old are also at a higher risk due to immature renal function.30
A number of other factors have been discussed as potentially contributing to the development of NSF. These include coexistent acidosis, erythropoietic stimulating agents (recombinant human erythropoietin), major proinflammatory events (major surgery, infection/sepsis, vascular events, or thrombosis), a hypercoagulable state, hyperphosphatemia, medications that could cause transmetallation of gadolinium (iron, zinc, copper, calcium, lanthanum, and sevelamer) and concomitant immunosuppression.31-33 While such conditions and circumstances have been noted in various reports, these have not been found to be consistently reproducible.
Clinical Features
The clinical findings in NSF can be quite inconspicuous in the early stages, yet they may progress to pronounced cutaneous manifestations in the late phases. Initially, patients typically complain of pruritus and/or pain, which have been described as numbness, tingling, burning, sharp, and sizzling.34 In well-developed NSF, patients can have joint contractures and report loss in range of motion with limited mobility.
On physical examination, the predominant sites of involvement are the bilateral lower and upper extremities. The trunk can be affected as well; however, the face is characteristically spared. Early lesions may present as subtle superficial papules and plaques and/or focal areas of hair loss (Figure 1). Scattered depigmented scar-like macules can also be seen (Figure 2). Edema of the lower extremities is commonly noted, which is increased over that found at baseline for many of the patients with chronic renal failure. Lesions of longer duration show deep dermal and subcutaneous induration, resulting in a hardened woody consistency (Figures 3a and 3b). Girardi recently described a phenomenon noted in his NSF patients that he termed “banding” whereby the underlying fibrosis pulls the skin down in a linear band.34 The overlying skin can have irregular erythema or velvety discoloration and often becomes dimpled, imparting a cobblestone appearance likened to the skin of an orange (peau d’ orange) (Figure 4). In the most severe forms, patients develop severe disabling joint contractures.
Ocular lesions have been reported, which take the form of whitish-yellow scleral plaques with vascular telangiectasias.35 Visceral involvement is variable; however, fibrosis has been documented in the heart, lungs, liver, kidneys, testes, and skeletal muscles.3,36,37
Histopathological Features
Microscopic examination of cutaneous lesional biopsies shows dermal and subcutaneous fibrosis. The thickened collagen bundles primarily occupy the mid and lower dermis with widening of the subcutaneous septa (Figure 5). Occasionally, there is extension into the underlying fascia or skeletal muscle. An increased deposition of dermal mucin and elastic fibers is seen. In addition, there is a proliferation of histiocytes and dermal dendrocytes, including multinucleated forms. The characteristic finding, however, is the presence of bland non-mitotically active spindle cells scattered within the fibrotic background (Figure 6).
Special and immunohistochemical stains can assist in highlighting these features. Stains for mucin, such as colloidal iron, will confirm the increase in dermal mucin (Figure 7). An elastic stain, such as Verhoff-van Gieson, can demonstrate thickened and more pronounced elastic tissue fibers within the dermis. A CD34 immunohistochemical stain will decorate the dermal spindle cell proliferation. However, if confirmation of these cells as circulating fibrocytes is sought, dual immunohistochemical staining with CD34 and procollagen will be positive. The augmentation of histiocytes and dermal dendrocytes can be established by positive staining with CD68 and Factor XIIIa.
Differential Diagnosis
The differential diagnosis of similar fibrosing cutaneous conditions mainly includes scleromyxedema, systemic sclerosis/morphea, eosinophilic fasciitis, and lipodermatosclerosis.
Scleromyxedema. Histologically, differentiating NSF from scleromyxedema can be the most challenging. A 2005 paper by Kucher et al focused on just that.38 The authors concluded that there were no pathognomonic microscopic features to reliably distinguish between the two entities, either on hematoxylin and eosin examination or by immunohistochemical staining profiles.
Therefore, the mainstay and most accurate way to separate NSF from scleromyxedema are by clinical history and findings. Patients with scleromyxedema have systemic manifestations, most notably a serum monoclonal paraprotein as well as prominent mucin deposition in numerous visceral organs. The cutaneous lesions have a predilection for the arms, hands, and trunk and classically involve the face, which has been universally spared thus far in all reported cases of NSF.
Systemic sclerosis/morphea is an autoimmune connective tissue disorder with similar clinical manifestations of symmetrical hardening of the skin. In addition to cutaneous involvement, the systemic disease (systemic sclerosis) affects the blood vessels and visceral organs, including the gastrointestinal tract, heart and kidneys.
Positive serum markers include antinuclear antibody, anticentromere antibody, and Scl-70. The localized form (morphea) is limited to the skin and presents with indurated plaques on the trunk and extremities. The histology differs in that there is a decrease in dermal spindle cells and elastic fibers as well as the consistent presence of plasma cells.
Eosinophilic fasciitis is another entity resulting in hardened woody induration of the skin with systemic evidence. First described by Shulman in 1974, this syndrome is characterized by fasciitis and fibrosis of the extremities (but with sparing of the hands and feet), polyclonal hypergammaglobulinemia, and peripheral eosinophilia.39
Under the microscope, the thickened collagen bundles layer through the subcutaneous tissue in a roughly parallel fashion with involvement of the underlying fascia and subfascial muscle. Conspicuous eosinophils are typically noted within the accompanying inflammatory infiltrate. Increased dermal mucin is not a feature.
Lipodermatosclerosis, also known as sclerosing panniculitis, is a chronic inflammatory and fibrosing condition of the bilateral lower extremities, classically occurring in obese adult females with a history of venous insufficiency.
The clinical appearance is of erythematous indurated painful woody plaques confined to the distal lower extremities.
The histology is quite dissimilar from that seen in NSF and is typified by exclusive involvement of lobular and septal subcutaneous tissue with fibrosis, fat necrosis, macro- and microcyst formation and lipomembranous change.
Treatment
Numerous forms of therapy have been tried with variable degrees of success. Partial or complete spontaneous remission has occurred, especially with resolution of the underlying renal impairment. Treatments range from supportive measures with physical therapy and pain management to more aggressive regimens, including extracorporeal photophoresis, high-dose intravenous immunoglobulin, immunostimulants (interferon-α), immunosuppressants (cyclosporine, methotrexate, mycofenolate mofetil, and oral and topical corticosteroids), pentoxifylline, thalidomide, photodynamic therapy, psoralen combined with UVA therapy, intravenous sodium thiosulfate, and ACE- inhibitors.12,18,19,26,40-41
Prevention
The most important and effective modality is prevention. Much of the recent research and literature has been dedicated to this crucial aspect. Some of the suggestions have been use of less toxic or implicated gadolinium-containing contrast agents, switch to alternative imaging procedures that avoid or limit exposure to gadolinium, giving the lowest possible dose and avoiding repeat administration when gadolinium-containing contrast agents are absolutely necessary, initiating hemodialysis after gadolinium-requiring imaging procedures to expedite rapid clearance, optimizing patient metabolic panels and treating underlying inflammatory or thrombotic conditions prior to injection of gadolinium, and avoidance of drugs that increase transglutaminases or TGF-β or that could be involved in transmetallation.15,19,23,32,42
Prognosis
The fibrosis in NSF can be rapidly progressive or quiescent and stable. Spontaneous remission has been known to occur. However, there is significant morbidity associated with well-developed cases, mainly as a result of loss in range of motion and mobility that accompanies disabling joint contractures. Systemic involvement can cause fibrosis in numerous visceral organs, leading to severe impairment and dire consequences. An increased mortality rate is associated with the disease, ranging from 7.7% to 67%.19
Conclusion
Nephrogenic systemic fibrosis is a debilitating disease occurring in patients with renal dysfunction that is relatively new to the medical community. Cutaneous involvement is inevitably present, with systemic complications being less frequent. While the details regarding the risk factors, pathogenesis, and effective treatment are still being explored, awareness and recognition of this entity is important in order to take the appropriate measures to prevent disease initiation or progression.
Dr. Walsh is a Dermatopathology Fellow, Departments of Dermatology and Pathology, Wake Forest University School of Medicine, Winston-Salem, NC.
Dr. Sangüeza is Professor, Departments of Dermatology and Pathology and Director of Dermatopathology, Wake Forest University School of Medicine, Winston-Salem, NC.
DISCLOSURE: Dr. Walsh and Dr. Sangüeza have no conflict of interest with any subjects discussed in this article.
CME #135 June 2008
Skin & Aging is proud to bring you this latest installment in its CME series. This series consists of regular CME activities that qualify you for 1 AMA PRA Category 1 Credit. As a reader of Skin & Aging, this course is brought to you free of charge — you aren’t required to pay a processing fee. In this CME, Drs. Sarah Walsh and Omar Sangüeza discuss nephrogenic systemic fibrosis (NSF), an emerging fibrosing condition that primarily affects the skin and has recently been linked to exposure to gadolinium-containing contrast agents. At the end of this article, you’ll find an exam. Mark your responses in the designated area, then fax page 41 to NACCME at (610) 560-0501.
We’ll also post this course on our Web site, which you can access at www.skinandaging.com. I hope this CME contributes to your clinical skills.
Amy McMichael, M.D. CME Editor
Amy McMichael, M.D., is Associate Professor in the Department of Dermatology, Director of the Hair Disorders Clinic and Residency Program Director at Wake Forest University Medical Center in Winston-Salem, NC.
Nephrogenic Systemic Fibrosis: A Comprehensive Review
By Sarah N. Walsh, M.D., and Omar P. Sangüeza, M.D.
Nephrogenic systemic fibrosis (NSF), initially referred to as nephrogenic fibrosing dermopathy (NFD), is an emerging fibrosing condition that primarily affects the skin and occurs in patients with renal injury. Recently, this disease has been linked to exposure to gadolinium-containing contrast agents. The disease can have a rapid or slowly progressive course, which generally begins with pain, pruritus, and swelling of the extremities and trunk and evolves to hardened woody induration with joint contractures, with or without systemic involvement. While partial or complete spontaneous remission has been reported, numerous other therapies have been tried with varying degrees of success. To date, prevention appears to be the most effective option. Significant morbidity and an increased mortality are the most pressing concerns, leading to continuous active research to further identify risk factors, inciting agents, pathogenesis, and effective treatment modalities.
Historical Perspective
In a Research Letter to the Lancet in 2000, Cowper et al reported on 15 renal dialysis patients who had a new cutaneous fibrosing disorder.1 It was recognized to be similar to scleromyxedema; however, the main distinguishing features were: 1. absence of a serum monoclonal paraprotein; 2. lack of systemic mucin deposition; and 3. sparing of the face. Shortly thereafter, it was again Cowper et al who wrote the seminal article “Nephrogenic Fibrosing Dermopathy,” which put a name to this new entity.2 This 2001 paper described indurated plaques and papules that developed on the trunk and extremities of patients with renal dysfunction. Histologically, the biopsies showed thickened dermal collagen with widening of subcutaneous septa and increased dermal mucin and elastic fibers. Stellate spindled cells that were CD34 positive were intermingled among the large dermal collagen bundles. Only one case in the series showed spontaneous resolution after dialysis for acute tubular necrosis.
In 2003, an explosion of published articles contributed to a wider understanding of NFD. In July, Ting et al reported on the first autopsy of a patient with NFD.3 This was significant as it provided the first undisputed evidence of systemic involvement in which extensive fibrosis and calcification were noted in the diaphragm, psoas muscle, renal tubules, and rete testes.
A Letter to the Editor by Cowper and Bucala in the American Journal of Dermatopathology in August hypothesized on the culprit cell of NFD.4 By using a dual immunohistochemical staining technique with CD34 and procollagen, they found that the majority of the dermal spindle cells were positive for both markers. These cells were thereby identified as circulating fibrocytes, which were first described in the rheumatology literature in 1994 and known to be bone marrow-derived cells that circulate as fibroblast progenitor cells, produce collagen, and aid in wound repair and remodeling.5
Since the majority of the reported cases to this point had occurred in patients receiving dialysis for severe renal disease, in 2004 Jimenez et al proposed a new terminology for the disorder, dialysis-associated systemic fibrosis (DASF).6 However, concurrent and subsequent reports demonstrated that this disease affects patients with a wide spectrum of acute and chronic renal disorders that did not necessarily require dialysis. In addition, further evidence supported systemic involvement. Therefore in 2005, the name of the disease was more accurately changed to nephrogenic systemic fibrosis.7
Gadolinium is a naturally occurring earth element that is a part of the lanthanoid series (atomic #64).9 This silvery white metal has seven unpaired electrons, which make it highly paramagnetic and therefore ideal to enhance images in magnetic resonance (MR) scanning. In 1988, gadolinium-based contrast agents were first used in humans.8 The use of these agents increased, and in 1997, the first paper implementing their utility in renal patients was published.10
While Cowper et al speculated that a “recently introduced material, possibly a contrast agent . . .” stimulated circulating fibrocytes, culminating in the fibrotic changes, it was Grobner et al who, in 2006, provided evidence of the first real connection between gadolinium and NSF.11,12 This connection was solidified with subsequent studies in which gadolinium was detected within tissues of patients with NSF.13
As evidence for this association accumulated, the U.S. Food and Drug Administration responded by issuing a public health advisory warning on the use of gadolinium-based contrast agents in patients with renal dysfunction.14
The 2007 guidelines of the European Society of Urogenital Radiology stated that “gadodiamide [the generic name for Omniscan] should not be administered to patients with reduced kidney function or to those on dialysis.”15
In addition to the discovery of the link between gadolinium and NSF in 2006, this year was also significant for publications that documented cases in other parts of the world — including Europe and India — as well as identification of the disease in children.16,17
The most recent reports have focused on other associations or risk factors for the development of NSF as well as effective treatment options.
Epidemiology
Yale researchers have maintained a registry called the International Center for Nephrogenic Fibrosing Dermopathy Research, which has identified more than 215 cases thus far (last updated Jan. 20, 2008).18 This condition seems to occur in approximately 4% of patients with advanced or end-stage renal disease.19 Patients have ranged in age from 8 to 86 years.20 Males and females are equally affected (M:F ratio of 1:1), and no racial predilection has been noted.20
Pathogenesis
The circulating fibrocyte has been implicated in the development of NSF. These cells have features of both connective tissue cells (procollagen-positive) and bone marrow derived progenitor cells (CD34-positive), hence the name fibrocyte, which combines fibroblast with leukocyte/thrombocyte/erythrocyte.21
Circulating fibrocytes produce chemokines and growth factors, including transforming growth factor-beta (TGF-β), which induces connective tissue matrix production as well as angiogenesis. Since these cells are not noted to be mitotically active on microscopic examination, it is speculated that they are recruited from the circulation, deposit collagen, and eventually mature into fibroblasts or myofibroblasts, at which point they lose their CD34 phenotype.21
The link between NSF and gadolinium-based contrast agents is now well established. Clearance of gadolinium is significantly reduced in the setting of renal dysfunction. Gadolinium has been detected in the insoluble unchelated toxic form (Gd3+) by quantitative scanning electron microscopy/energy-dispersive X-ray spectroscopy in the tissues of patients with NSF.22 In addition, higher dermal concentrations of gadolinium have been found in late lesions when compared with biopsies taken at the onset of the disease; this is thought to be from initial bone storage with subsequent mobilization.22 This free (unchelated) toxic Gd3+ is postulated to act as a target or trigger for recruitment of circulating fibrocytes.12
A recent article by Parsons et al has suggested another possible trigger in the pathogenesis of NSF.23 Through immunohistochemical staining methods, these authors found an increased expression of transglutaminases in biopsy specimens with the diagnosis of NSF. Gadolinium has been shown to activate transglutaminases, and transglutaminases in turn are known to activate TGF-β.24,25 As discussed previously, TGF-β induces fibroblastic proliferation.
While the exact stepwise development of the disease is still under investigation, renal dysfunction, gadolinium, circulating fibrocytes, TGF-β, and transglutaminases all seem to play a role, either in a dominant fashion or synergistically.
Risk Factors
Renal dysfunction is a prerequisite for NSF. It was once thought that only patients with severe renal disease — particularly those requiring dialysis — were affected. Subsequently, it became clear that any duration or degree of renal injury was sufficient. However, it has been well established that those at greatest risk are patients with chronic kidney disease or end-stage renal disease (ESRD) — particularly those with a glomerular filtration rate of <30 ml/min/1.73 m2 as well as those receiving peritoneal dialysis (even more so than hemodialysis).19
As highlighted previously, exposure to gadolinium is another monumental risk factor. While there have been reported cases of NSF occurring in patients without known exposure to the element, the vast majority have had at least one scan or procedure in which a gadolinium-based contrast agent was injected.26 The absolute risk for developing NSF in patients with ESRD has been reported as 2.4% per single MR study.27
The type of gadolinium-containing contrast agent used is also important. There are currently five agents approved by the FDA for use in humans. These differ mainly in relation to transmetallation and kinetic and thermodynamic stability (amount of free toxic Gd3+ released from the chelate by exchange with another metal).9 Gadodiamide (Omniscan) has a low thermodynamic stability and demonstrates more transmetallation, accounting for this agent being associated with the largest number of reported cases of NSF.9
There are additional factors to consider in relation to the use of gadolinium. Collidge et al found a positive association between the cumulative dose of gadolinium and the development of NSF.28 In patients with severe liver disease, the glomerular filtration rate is often overestimated, resulting in a greater risk of gadolinium exposure in this subgroup.29 Children less than 1 year old are also at a higher risk due to immature renal function.30
A number of other factors have been discussed as potentially contributing to the development of NSF. These include coexistent acidosis, erythropoietic stimulating agents (recombinant human erythropoietin), major proinflammatory events (major surgery, infection/sepsis, vascular events, or thrombosis), a hypercoagulable state, hyperphosphatemia, medications that could cause transmetallation of gadolinium (iron, zinc, copper, calcium, lanthanum, and sevelamer) and concomitant immunosuppression.31-33 While such conditions and circumstances have been noted in various reports, these have not been found to be consistently reproducible.
Clinical Features
The clinical findings in NSF can be quite inconspicuous in the early stages, yet they may progress to pronounced cutaneous manifestations in the late phases. Initially, patients typically complain of pruritus and/or pain, which have been described as numbness, tingling, burning, sharp, and sizzling.34 In well-developed NSF, patients can have joint contractures and report loss in range of motion with limited mobility.
On physical examination, the predominant sites of involvement are the bilateral lower and upper extremities. The trunk can be affected as well; however, the face is characteristically spared. Early lesions may present as subtle superficial papules and plaques and/or focal areas of hair loss (Figure 1). Scattered depigmented scar-like macules can also be seen (Figure 2). Edema of the lower extremities is commonly noted, which is increased over that found at baseline for many of the patients with chronic renal failure. Lesions of longer duration show deep dermal and subcutaneous induration, resulting in a hardened woody consistency (Figures 3a and 3b). Girardi recently described a phenomenon noted in his NSF patients that he termed “banding” whereby the underlying fibrosis pulls the skin down in a linear band.34 The overlying skin can have irregular erythema or velvety discoloration and often becomes dimpled, imparting a cobblestone appearance likened to the skin of an orange (peau d’ orange) (Figure 4). In the most severe forms, patients develop severe disabling joint contractures.
Ocular lesions have been reported, which take the form of whitish-yellow scleral plaques with vascular telangiectasias.35 Visceral involvement is variable; however, fibrosis has been documented in the heart, lungs, liver, kidneys, testes, and skeletal muscles.3,36,37
Histopathological Features
Microscopic examination of cutaneous lesional biopsies shows dermal and subcutaneous fibrosis. The thickened collagen bundles primarily occupy the mid and lower dermis with widening of the subcutaneous septa (Figure 5). Occasionally, there is extension into the underlying fascia or skeletal muscle. An increased deposition of dermal mucin and elastic fibers is seen. In addition, there is a proliferation of histiocytes and dermal dendrocytes, including multinucleated forms. The characteristic finding, however, is the presence of bland non-mitotically active spindle cells scattered within the fibrotic background (Figure 6).
Special and immunohistochemical stains can assist in highlighting these features. Stains for mucin, such as colloidal iron, will confirm the increase in dermal mucin (Figure 7). An elastic stain, such as Verhoff-van Gieson, can demonstrate thickened and more pronounced elastic tissue fibers within the dermis. A CD34 immunohistochemical stain will decorate the dermal spindle cell proliferation. However, if confirmation of these cells as circulating fibrocytes is sought, dual immunohistochemical staining with CD34 and procollagen will be positive. The augmentation of histiocytes and dermal dendrocytes can be established by positive staining with CD68 and Factor XIIIa.
Differential Diagnosis
The differential diagnosis of similar fibrosing cutaneous conditions mainly includes scleromyxedema, systemic sclerosis/morphea, eosinophilic fasciitis, and lipodermatosclerosis.
Scleromyxedema. Histologically, differentiating NSF from scleromyxedema can be the most challenging. A 2005 paper by Kucher et al focused on just that.38 The authors concluded that there were no pathognomonic microscopic features to reliably distinguish between the two entities, either on hematoxylin and eosin examination or by immunohistochemical staining profiles.
Therefore, the mainstay and most accurate way to separate NSF from scleromyxedema are by clinical history and findings. Patients with scleromyxedema have systemic manifestations, most notably a serum monoclonal paraprotein as well as prominent mucin deposition in numerous visceral organs. The cutaneous lesions have a predilection for the arms, hands, and trunk and classically involve the face, which has been universally spared thus far in all reported cases of NSF.
Systemic sclerosis/morphea is an autoimmune connective tissue disorder with similar clinical manifestations of symmetrical hardening of the skin. In addition to cutaneous involvement, the systemic disease (systemic sclerosis) affects the blood vessels and visceral organs, including the gastrointestinal tract, heart and kidneys.
Positive serum markers include antinuclear antibody, anticentromere antibody, and Scl-70. The localized form (morphea) is limited to the skin and presents with indurated plaques on the trunk and extremities. The histology differs in that there is a decrease in dermal spindle cells and elastic fibers as well as the consistent presence of plasma cells.
Eosinophilic fasciitis is another entity resulting in hardened woody induration of the skin with systemic evidence. First described by Shulman in 1974, this syndrome is characterized by fasciitis and fibrosis of the extremities (but with sparing of the hands and feet), polyclonal hypergammaglobulinemia, and peripheral eosinophilia.39
Under the microscope, the thickened collagen bundles layer through the subcutaneous tissue in a roughly parallel fashion with involvement of the underlying fascia and subfascial muscle. Conspicuous eosinophils are typically noted within the accompanying inflammatory infiltrate. Increased dermal mucin is not a feature.
Lipodermatosclerosis, also known as sclerosing panniculitis, is a chronic inflammatory and fibrosing condition of the bilateral lower extremities, classically occurring in obese adult females with a history of venous insufficiency.
The clinical appearance is of erythematous indurated painful woody plaques confined to the distal lower extremities.
The histology is quite dissimilar from that seen in NSF and is typified by exclusive involvement of lobular and septal subcutaneous tissue with fibrosis, fat necrosis, macro- and microcyst formation and lipomembranous change.
Treatment
Numerous forms of therapy have been tried with variable degrees of success. Partial or complete spontaneous remission has occurred, especially with resolution of the underlying renal impairment. Treatments range from supportive measures with physical therapy and pain management to more aggressive regimens, including extracorporeal photophoresis, high-dose intravenous immunoglobulin, immunostimulants (interferon-α), immunosuppressants (cyclosporine, methotrexate, mycofenolate mofetil, and oral and topical corticosteroids), pentoxifylline, thalidomide, photodynamic therapy, psoralen combined with UVA therapy, intravenous sodium thiosulfate, and ACE- inhibitors.12,18,19,26,40-41
Prevention
The most important and effective modality is prevention. Much of the recent research and literature has been dedicated to this crucial aspect. Some of the suggestions have been use of less toxic or implicated gadolinium-containing contrast agents, switch to alternative imaging procedures that avoid or limit exposure to gadolinium, giving the lowest possible dose and avoiding repeat administration when gadolinium-containing contrast agents are absolutely necessary, initiating hemodialysis after gadolinium-requiring imaging procedures to expedite rapid clearance, optimizing patient metabolic panels and treating underlying inflammatory or thrombotic conditions prior to injection of gadolinium, and avoidance of drugs that increase transglutaminases or TGF-β or that could be involved in transmetallation.15,19,23,32,42
Prognosis
The fibrosis in NSF can be rapidly progressive or quiescent and stable. Spontaneous remission has been known to occur. However, there is significant morbidity associated with well-developed cases, mainly as a result of loss in range of motion and mobility that accompanies disabling joint contractures. Systemic involvement can cause fibrosis in numerous visceral organs, leading to severe impairment and dire consequences. An increased mortality rate is associated with the disease, ranging from 7.7% to 67%.19
Conclusion
Nephrogenic systemic fibrosis is a debilitating disease occurring in patients with renal dysfunction that is relatively new to the medical community. Cutaneous involvement is inevitably present, with systemic complications being less frequent. While the details regarding the risk factors, pathogenesis, and effective treatment are still being explored, awareness and recognition of this entity is important in order to take the appropriate measures to prevent disease initiation or progression.
Dr. Walsh is a Dermatopathology Fellow, Departments of Dermatology and Pathology, Wake Forest University School of Medicine, Winston-Salem, NC.
Dr. Sangüeza is Professor, Departments of Dermatology and Pathology and Director of Dermatopathology, Wake Forest University School of Medicine, Winston-Salem, NC.
DISCLOSURE: Dr. Walsh and Dr. Sangüeza have no conflict of interest with any subjects discussed in this article.