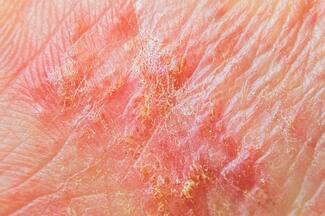
PATIENT PRESENTATION A 63-year-old African-American male presented with a 2-year history of pruritic nodules that started on the legs and spread to the trunk and arms. New lesions were associated with flares in pruritus. The patient has a past medical history of hypertension, hypercholesterolemia, stroke, myocardial infarction and stage 4 chronic kidney disease, with a glomerular filtration rate of 26 ml/min/1.73m2 (Normal ≥ 60ml/min/1.73m2). On physical examination, there were numerous follicular and perifollicular hyperkeratotic papules and nodules on the lower legs. Similar lesions were present on the abdomen and arms. Some lesions on the lower legs were arranged in a linear pattern, as shown in the Figure above. Numerous excoriations were seen on trunk and extremities. _________________________
Diagnosis: Acquired Perforating Dermatosis
Acquired perforating dermatosis (APD) is a disorder in which keratotic follicular or perifollicular papules develop, most frequently over extensor surfaces of the legs, but generalized lesions may be seen. It occurs most commonly in adults with renal failure or diabetes mellitus.1 The term Kyrle’s disease may be used synonymously with acquired perforating dermatosis.2 In 1916, Josef Kyrle (University of Vienna, Vienna, Austria) first reported on a diabetic woman with generalized hyperkeratotic nodules, which he called “hyperkeratosis follicularis et parafollicularis in cutem penetrans.3” This term emphasized that these hyperkeratotic perforating lesions were both follicular and perifollicular. Although APD can develop pre-dialysis, upon commencement of dialysis or post-dialysis; dialysis patients have a 10% prevalence rate.2 There are also a few reports of APD in patients with other systemic disorders, like cirrhosis or hepatocellular carcinoma,4,5 and even in those without any medical problems.6 APD is one of a larger group of disorders known as the perforating diseases, in which dermal connective tissue is eliminated through the epidermis. The exact pathogenesis remains to be elucidated. It is thought that the pruritus of renal disease may lead to chronic scratching via the accumulation of an unidentified, poorly dialysable, “uremic” substance. This may result in epithelial hyperplasia, possibly yielding classic perforating lesions.7 It has further been postulated that the metabolic derangements associated with chronic renal failure and diabetes mellitus may cause recognition of “foreign” substances within the connective tissue of the superficial dermis, which subsequently trigger a transepithelial elimination response.8 There may also be fragmentation of collagen or elastic fibers. More recent studies have proven that patients with diabetes mellitus and uremia have increased levels of fibronectin in the serum and at sites of perforating lesions in the skin. Therefore, fibronectin may be the specific “antigenic trigger,” and a candidate for pathogenesis of APD.9 Microangiopathy associated with diabetes, an imbalance in the expression of metalloproteinases and their tissue inhibitors — as well as deposition of uric acid, hydroxyapatite or silicon, abnormal vitamin A or D metabolism, and neutrophilic enzyme release — are all pathomechanisms that have been proposed.10,11,12 DIFFERENTIAL DIAGNOSIS The perforating disorders (Table 1) are composed of three major entities. These include reactive perforating collagenosis and elastosis perforans serpiginosa — both of which are inheritable — and acquired perforating dermatosis. Perforating folliculitis and perforating periumbilical calcific elastosis are also included in this group. When making the diagnosis of APD, one must consider the complete spectrum of perforating disorders. Differentiation is typically done based on factors such as incidence, time of onset, location, family history and perforating substances. On histology, APD can present as any of the other perforating diseases. Therefore, if an adult patient with underlying renal disease presents with hyperkeratotic lesions, APD should be considered, regardless of the perforating substance. Reactive perforating collagenosis (RPC) usually presents in childhood with keratotic papules 3 to 4 weeks following superficial trauma, usually on the arms and hands. Koebnerization is more commonly seen in RPC than in the other perforating disorders. Here, injury to the skin results in new lesions, usually in a linear distribution (as shown in the above photo). The papules of this rare familial disorder typically resolve spontaneously over 6 to 8 weeks, but in a very rare non-familial variant of RPC, severe trauma to the skin results in verrucous papules.14 Biopsy of RPC reveals transelimination of collagen within the plug of crusting or hyperkeratosis. Like RPC, elastosis perforans serpiginosa (EPS) is another rare disorder that usually begins during childhood. It has a strong association with genetic disorders such as Down syndrome, Marfan syndrome, acrogeria (premature aging syndrome with atrophy of skin and subcutaneous tissue), pseudoxanthoma elasticum, osteogenesis imperfecta, Rothmund-Thomson syndrome, Ehlers-Danlos syndrome and scleroderma.15 Additionally, drug- induced EPS may be caused by penicillamine.16 EPS lesions tend to be located on the lateral neck, face arms or flexural areas, and consist of 2 mm to 5 mm keratotic papules arranged in a serpiginous or annular pattern. The lesions may resolve spontaneously or persist for years. The majority of patients experience mild pruritus or no symptoms at all. Biopsy of EPS lesions is similar to that of RPC, but elastic fibers are visualized in the plug or within the epidermis. Some authors describe a fourth type of perforating disorder known as “perforating folliculitis.” However, others suggest that this is not a specific entity, because rupture of the follicle may occur in any type of folliculitis, regardless of the etiology.7 It is important to note that small lesions of APD often have the histologic picture of perforating folliculitis. In addition to the perforating diseases, other disorders characterized by papules or nodules with central keratotic plugs or crusts should be included in the differential diagnosis. These include, but are not limited to, prurigo nodularis, phrynoderma, perforating of exogenous or endogenous substances, eruptive keratoacanthomas, hypertrophic lichen planus, dermatofibromas and arthropod bites. Many patients with APD may have coexisting folliculitis and prurigo nodularis. Therefore, it is important to realize that one patient may have multiple different types of lesions at different stages of development and that a biopsy of just one lesion may not be representative of the entire process. The Koebner phenomenon with lesions in a linear arrangement is most commonly observed in RPC but can occasionally occur in APD17 or any of the other perforating disorders. Occurrence of the Koebner phenomenon should also bring to mind psoriasis, hypertrophic lichen planus and verruca as differential diagnoses. HISTOLOGY All of the perforating diseases consist of a plug of crusting or hyperkeratosis, with parakeratosis. It is important to examine sections from many levels through the tissue in order to find the site of perforation. The aggregating cell type in perforating diseases also varies. Older lesions tend to have lymphocytes, macrophages or multinucleated giant cells at the perforation site in the dermis. Other lesions may reveal neutrophilic aggregates in the dermis. The Verhoeff-van Gieson (VVG) stain is particularly useful in histological identification of perforating diseases; the stain gives a black appearance to elastic fibers and a red appearance to collagen fibers. (See Figure 1.) Identification of the specific perforating substance is key to differentiating the various perforating disorders. In RPC, there is transepidermal elimination of collagen fibers, whereas the perforating substance in EPS is elastic fibers. Biopsy findings in acquired perforating dermatosis are particularly variable. The histologic finding of transepidermal elimination of both collagen and elastic fibers in patients with hyperkeratotic papules and a history of renal disease or diabetes mellitus was important in the acceptance of acquired perforating dermatosis as a distinct disease entity amongst other perforating disorders.1 Histologic findings in APD can also be identical to perforating folliculitis, childhood EPS, RPC or may be very non-specific with amorphous degenerated material within perforations.6 When APD presents with perforating folliculitis, the perforation may be in the infundibular portion of the hair follicle.18,19 In larger lesions, the perforations may be at the base of the follicular invagination.20,21 MANAGEMENT As pruritus is the most common symptom of APD, antihistamines and other antipruritic agents do provide limited relief. Narrowband UVB phototherapy is the most effective treatment option for patients with APD and renal disease, as it relieves coexisting pruritus.22 Topical, intralesional or systemic corticosteroids or retinoid therapy have been associated with clinical clearance or significant improvement of lesions.2 There are also anecdotal reports of successful treatment with topical menthol, salicylic acid, sulfur and benzoyl peroxide;7 systemic antibiotics such as doxycycline and clindamycin;23,24 and allopurinol in patients with elevated serum uric acid levels.25 Renal transplantation has been associated with cure in occasional patients with dialysis-associated APD.11 Our Patient Our patient’s biopsy was consistent with perforating folliculitis. There was hyperkeratosis and parakeratosis, and a neutrophilic infiltrate within, and perforating the follicular epithelium. (See Figure 2.) The VVG stain showed amorphous degenerated material within the follicle, which is not clearly identifiable as collagen or elastic fibers. Our patient was treated with antihistamines, topical corticosteroids and narrowband UVB phototherapy and noted minimal relief of pruritus. On his last visit in June 2009, he opted to discontinue NBUVB, as new lesions were still occurring on the abdomen and legs. CONCLUSION Acquired perforating dermatosis is a distinct entity within the scope of perforating diseases. It presents with keratotic follicular or perifollicular papules, most frequently over extensor surfaces of the legs. It is important to note that histologically, a lesion may resemble any of the other perforating disorders; or may simply consist of transelimination of necrotic material. A spectrum of different lesions may also be found on a single patient. Therefore, this is a largely clinical diagnosis; and in an adult patient with underlying renal disease or diabetes mellitus, APD should be strongly considered. The exact etiology of APD is unknown, and continuing research may formally identify fibronectin or possibly another pruritogenic factor as the culprit. Early recognition of the signs, symptoms and disease associations in APD can facilitate rapid diagnosis and initiation of appropriate treatment. Dr. Rodney is a dermatology resident in the Department of Dermatology, Howard University Hospital and Veterans Affairs Medical Center, Washington DC. Charla S. Taylor is a medical student at Howard University College of Medicine, Washington DC. Dr. Cohen is former Chief, Department of Dermatology, Veterans Affair Medical Center, Washington DC. Dr. Khachemoune, the Section Editor of Derm Dx, is with Department of Dermatology, State University of New York, Brooklyn, NY. Disclosure: The authors have no conflict of interest with any material presented in this column.
PATIENT PRESENTATION A 63-year-old African-American male presented with a 2-year history of pruritic nodules that started on the legs and spread to the trunk and arms. New lesions were associated with flares in pruritus. The patient has a past medical history of hypertension, hypercholesterolemia, stroke, myocardial infarction and stage 4 chronic kidney disease, with a glomerular filtration rate of 26 ml/min/1.73m2 (Normal ≥ 60ml/min/1.73m2). On physical examination, there were numerous follicular and perifollicular hyperkeratotic papules and nodules on the lower legs. Similar lesions were present on the abdomen and arms. Some lesions on the lower legs were arranged in a linear pattern, as shown in the Figure above. Numerous excoriations were seen on trunk and extremities. _________________________
Diagnosis: Acquired Perforating Dermatosis
Acquired perforating dermatosis (APD) is a disorder in which keratotic follicular or perifollicular papules develop, most frequently over extensor surfaces of the legs, but generalized lesions may be seen. It occurs most commonly in adults with renal failure or diabetes mellitus.1 The term Kyrle’s disease may be used synonymously with acquired perforating dermatosis.2 In 1916, Josef Kyrle (University of Vienna, Vienna, Austria) first reported on a diabetic woman with generalized hyperkeratotic nodules, which he called “hyperkeratosis follicularis et parafollicularis in cutem penetrans.3” This term emphasized that these hyperkeratotic perforating lesions were both follicular and perifollicular. Although APD can develop pre-dialysis, upon commencement of dialysis or post-dialysis; dialysis patients have a 10% prevalence rate.2 There are also a few reports of APD in patients with other systemic disorders, like cirrhosis or hepatocellular carcinoma,4,5 and even in those without any medical problems.6 APD is one of a larger group of disorders known as the perforating diseases, in which dermal connective tissue is eliminated through the epidermis. The exact pathogenesis remains to be elucidated. It is thought that the pruritus of renal disease may lead to chronic scratching via the accumulation of an unidentified, poorly dialysable, “uremic” substance. This may result in epithelial hyperplasia, possibly yielding classic perforating lesions.7 It has further been postulated that the metabolic derangements associated with chronic renal failure and diabetes mellitus may cause recognition of “foreign” substances within the connective tissue of the superficial dermis, which subsequently trigger a transepithelial elimination response.8 There may also be fragmentation of collagen or elastic fibers. More recent studies have proven that patients with diabetes mellitus and uremia have increased levels of fibronectin in the serum and at sites of perforating lesions in the skin. Therefore, fibronectin may be the specific “antigenic trigger,” and a candidate for pathogenesis of APD.9 Microangiopathy associated with diabetes, an imbalance in the expression of metalloproteinases and their tissue inhibitors — as well as deposition of uric acid, hydroxyapatite or silicon, abnormal vitamin A or D metabolism, and neutrophilic enzyme release — are all pathomechanisms that have been proposed.10,11,12 DIFFERENTIAL DIAGNOSIS The perforating disorders (Table 1) are composed of three major entities. These include reactive perforating collagenosis and elastosis perforans serpiginosa — both of which are inheritable — and acquired perforating dermatosis. Perforating folliculitis and perforating periumbilical calcific elastosis are also included in this group. When making the diagnosis of APD, one must consider the complete spectrum of perforating disorders. Differentiation is typically done based on factors such as incidence, time of onset, location, family history and perforating substances. On histology, APD can present as any of the other perforating diseases. Therefore, if an adult patient with underlying renal disease presents with hyperkeratotic lesions, APD should be considered, regardless of the perforating substance. Reactive perforating collagenosis (RPC) usually presents in childhood with keratotic papules 3 to 4 weeks following superficial trauma, usually on the arms and hands. Koebnerization is more commonly seen in RPC than in the other perforating disorders. Here, injury to the skin results in new lesions, usually in a linear distribution (as shown in the above photo). The papules of this rare familial disorder typically resolve spontaneously over 6 to 8 weeks, but in a very rare non-familial variant of RPC, severe trauma to the skin results in verrucous papules.14 Biopsy of RPC reveals transelimination of collagen within the plug of crusting or hyperkeratosis. Like RPC, elastosis perforans serpiginosa (EPS) is another rare disorder that usually begins during childhood. It has a strong association with genetic disorders such as Down syndrome, Marfan syndrome, acrogeria (premature aging syndrome with atrophy of skin and subcutaneous tissue), pseudoxanthoma elasticum, osteogenesis imperfecta, Rothmund-Thomson syndrome, Ehlers-Danlos syndrome and scleroderma.15 Additionally, drug- induced EPS may be caused by penicillamine.16 EPS lesions tend to be located on the lateral neck, face arms or flexural areas, and consist of 2 mm to 5 mm keratotic papules arranged in a serpiginous or annular pattern. The lesions may resolve spontaneously or persist for years. The majority of patients experience mild pruritus or no symptoms at all. Biopsy of EPS lesions is similar to that of RPC, but elastic fibers are visualized in the plug or within the epidermis. Some authors describe a fourth type of perforating disorder known as “perforating folliculitis.” However, others suggest that this is not a specific entity, because rupture of the follicle may occur in any type of folliculitis, regardless of the etiology.7 It is important to note that small lesions of APD often have the histologic picture of perforating folliculitis. In addition to the perforating diseases, other disorders characterized by papules or nodules with central keratotic plugs or crusts should be included in the differential diagnosis. These include, but are not limited to, prurigo nodularis, phrynoderma, perforating of exogenous or endogenous substances, eruptive keratoacanthomas, hypertrophic lichen planus, dermatofibromas and arthropod bites. Many patients with APD may have coexisting folliculitis and prurigo nodularis. Therefore, it is important to realize that one patient may have multiple different types of lesions at different stages of development and that a biopsy of just one lesion may not be representative of the entire process. The Koebner phenomenon with lesions in a linear arrangement is most commonly observed in RPC but can occasionally occur in APD17 or any of the other perforating disorders. Occurrence of the Koebner phenomenon should also bring to mind psoriasis, hypertrophic lichen planus and verruca as differential diagnoses. HISTOLOGY All of the perforating diseases consist of a plug of crusting or hyperkeratosis, with parakeratosis. It is important to examine sections from many levels through the tissue in order to find the site of perforation. The aggregating cell type in perforating diseases also varies. Older lesions tend to have lymphocytes, macrophages or multinucleated giant cells at the perforation site in the dermis. Other lesions may reveal neutrophilic aggregates in the dermis. The Verhoeff-van Gieson (VVG) stain is particularly useful in histological identification of perforating diseases; the stain gives a black appearance to elastic fibers and a red appearance to collagen fibers. (See Figure 1.) Identification of the specific perforating substance is key to differentiating the various perforating disorders. In RPC, there is transepidermal elimination of collagen fibers, whereas the perforating substance in EPS is elastic fibers. Biopsy findings in acquired perforating dermatosis are particularly variable. The histologic finding of transepidermal elimination of both collagen and elastic fibers in patients with hyperkeratotic papules and a history of renal disease or diabetes mellitus was important in the acceptance of acquired perforating dermatosis as a distinct disease entity amongst other perforating disorders.1 Histologic findings in APD can also be identical to perforating folliculitis, childhood EPS, RPC or may be very non-specific with amorphous degenerated material within perforations.6 When APD presents with perforating folliculitis, the perforation may be in the infundibular portion of the hair follicle.18,19 In larger lesions, the perforations may be at the base of the follicular invagination.20,21 MANAGEMENT As pruritus is the most common symptom of APD, antihistamines and other antipruritic agents do provide limited relief. Narrowband UVB phototherapy is the most effective treatment option for patients with APD and renal disease, as it relieves coexisting pruritus.22 Topical, intralesional or systemic corticosteroids or retinoid therapy have been associated with clinical clearance or significant improvement of lesions.2 There are also anecdotal reports of successful treatment with topical menthol, salicylic acid, sulfur and benzoyl peroxide;7 systemic antibiotics such as doxycycline and clindamycin;23,24 and allopurinol in patients with elevated serum uric acid levels.25 Renal transplantation has been associated with cure in occasional patients with dialysis-associated APD.11 Our Patient Our patient’s biopsy was consistent with perforating folliculitis. There was hyperkeratosis and parakeratosis, and a neutrophilic infiltrate within, and perforating the follicular epithelium. (See Figure 2.) The VVG stain showed amorphous degenerated material within the follicle, which is not clearly identifiable as collagen or elastic fibers. Our patient was treated with antihistamines, topical corticosteroids and narrowband UVB phototherapy and noted minimal relief of pruritus. On his last visit in June 2009, he opted to discontinue NBUVB, as new lesions were still occurring on the abdomen and legs. CONCLUSION Acquired perforating dermatosis is a distinct entity within the scope of perforating diseases. It presents with keratotic follicular or perifollicular papules, most frequently over extensor surfaces of the legs. It is important to note that histologically, a lesion may resemble any of the other perforating disorders; or may simply consist of transelimination of necrotic material. A spectrum of different lesions may also be found on a single patient. Therefore, this is a largely clinical diagnosis; and in an adult patient with underlying renal disease or diabetes mellitus, APD should be strongly considered. The exact etiology of APD is unknown, and continuing research may formally identify fibronectin or possibly another pruritogenic factor as the culprit. Early recognition of the signs, symptoms and disease associations in APD can facilitate rapid diagnosis and initiation of appropriate treatment. Dr. Rodney is a dermatology resident in the Department of Dermatology, Howard University Hospital and Veterans Affairs Medical Center, Washington DC. Charla S. Taylor is a medical student at Howard University College of Medicine, Washington DC. Dr. Cohen is former Chief, Department of Dermatology, Veterans Affair Medical Center, Washington DC. Dr. Khachemoune, the Section Editor of Derm Dx, is with Department of Dermatology, State University of New York, Brooklyn, NY. Disclosure: The authors have no conflict of interest with any material presented in this column.
PATIENT PRESENTATION A 63-year-old African-American male presented with a 2-year history of pruritic nodules that started on the legs and spread to the trunk and arms. New lesions were associated with flares in pruritus. The patient has a past medical history of hypertension, hypercholesterolemia, stroke, myocardial infarction and stage 4 chronic kidney disease, with a glomerular filtration rate of 26 ml/min/1.73m2 (Normal ≥ 60ml/min/1.73m2). On physical examination, there were numerous follicular and perifollicular hyperkeratotic papules and nodules on the lower legs. Similar lesions were present on the abdomen and arms. Some lesions on the lower legs were arranged in a linear pattern, as shown in the Figure above. Numerous excoriations were seen on trunk and extremities. _________________________
Diagnosis: Acquired Perforating Dermatosis
Acquired perforating dermatosis (APD) is a disorder in which keratotic follicular or perifollicular papules develop, most frequently over extensor surfaces of the legs, but generalized lesions may be seen. It occurs most commonly in adults with renal failure or diabetes mellitus.1 The term Kyrle’s disease may be used synonymously with acquired perforating dermatosis.2 In 1916, Josef Kyrle (University of Vienna, Vienna, Austria) first reported on a diabetic woman with generalized hyperkeratotic nodules, which he called “hyperkeratosis follicularis et parafollicularis in cutem penetrans.3” This term emphasized that these hyperkeratotic perforating lesions were both follicular and perifollicular. Although APD can develop pre-dialysis, upon commencement of dialysis or post-dialysis; dialysis patients have a 10% prevalence rate.2 There are also a few reports of APD in patients with other systemic disorders, like cirrhosis or hepatocellular carcinoma,4,5 and even in those without any medical problems.6 APD is one of a larger group of disorders known as the perforating diseases, in which dermal connective tissue is eliminated through the epidermis. The exact pathogenesis remains to be elucidated. It is thought that the pruritus of renal disease may lead to chronic scratching via the accumulation of an unidentified, poorly dialysable, “uremic” substance. This may result in epithelial hyperplasia, possibly yielding classic perforating lesions.7 It has further been postulated that the metabolic derangements associated with chronic renal failure and diabetes mellitus may cause recognition of “foreign” substances within the connective tissue of the superficial dermis, which subsequently trigger a transepithelial elimination response.8 There may also be fragmentation of collagen or elastic fibers. More recent studies have proven that patients with diabetes mellitus and uremia have increased levels of fibronectin in the serum and at sites of perforating lesions in the skin. Therefore, fibronectin may be the specific “antigenic trigger,” and a candidate for pathogenesis of APD.9 Microangiopathy associated with diabetes, an imbalance in the expression of metalloproteinases and their tissue inhibitors — as well as deposition of uric acid, hydroxyapatite or silicon, abnormal vitamin A or D metabolism, and neutrophilic enzyme release — are all pathomechanisms that have been proposed.10,11,12 DIFFERENTIAL DIAGNOSIS The perforating disorders (Table 1) are composed of three major entities. These include reactive perforating collagenosis and elastosis perforans serpiginosa — both of which are inheritable — and acquired perforating dermatosis. Perforating folliculitis and perforating periumbilical calcific elastosis are also included in this group. When making the diagnosis of APD, one must consider the complete spectrum of perforating disorders. Differentiation is typically done based on factors such as incidence, time of onset, location, family history and perforating substances. On histology, APD can present as any of the other perforating diseases. Therefore, if an adult patient with underlying renal disease presents with hyperkeratotic lesions, APD should be considered, regardless of the perforating substance. Reactive perforating collagenosis (RPC) usually presents in childhood with keratotic papules 3 to 4 weeks following superficial trauma, usually on the arms and hands. Koebnerization is more commonly seen in RPC than in the other perforating disorders. Here, injury to the skin results in new lesions, usually in a linear distribution (as shown in the above photo). The papules of this rare familial disorder typically resolve spontaneously over 6 to 8 weeks, but in a very rare non-familial variant of RPC, severe trauma to the skin results in verrucous papules.14 Biopsy of RPC reveals transelimination of collagen within the plug of crusting or hyperkeratosis. Like RPC, elastosis perforans serpiginosa (EPS) is another rare disorder that usually begins during childhood. It has a strong association with genetic disorders such as Down syndrome, Marfan syndrome, acrogeria (premature aging syndrome with atrophy of skin and subcutaneous tissue), pseudoxanthoma elasticum, osteogenesis imperfecta, Rothmund-Thomson syndrome, Ehlers-Danlos syndrome and scleroderma.15 Additionally, drug- induced EPS may be caused by penicillamine.16 EPS lesions tend to be located on the lateral neck, face arms or flexural areas, and consist of 2 mm to 5 mm keratotic papules arranged in a serpiginous or annular pattern. The lesions may resolve spontaneously or persist for years. The majority of patients experience mild pruritus or no symptoms at all. Biopsy of EPS lesions is similar to that of RPC, but elastic fibers are visualized in the plug or within the epidermis. Some authors describe a fourth type of perforating disorder known as “perforating folliculitis.” However, others suggest that this is not a specific entity, because rupture of the follicle may occur in any type of folliculitis, regardless of the etiology.7 It is important to note that small lesions of APD often have the histologic picture of perforating folliculitis. In addition to the perforating diseases, other disorders characterized by papules or nodules with central keratotic plugs or crusts should be included in the differential diagnosis. These include, but are not limited to, prurigo nodularis, phrynoderma, perforating of exogenous or endogenous substances, eruptive keratoacanthomas, hypertrophic lichen planus, dermatofibromas and arthropod bites. Many patients with APD may have coexisting folliculitis and prurigo nodularis. Therefore, it is important to realize that one patient may have multiple different types of lesions at different stages of development and that a biopsy of just one lesion may not be representative of the entire process. The Koebner phenomenon with lesions in a linear arrangement is most commonly observed in RPC but can occasionally occur in APD17 or any of the other perforating disorders. Occurrence of the Koebner phenomenon should also bring to mind psoriasis, hypertrophic lichen planus and verruca as differential diagnoses. HISTOLOGY All of the perforating diseases consist of a plug of crusting or hyperkeratosis, with parakeratosis. It is important to examine sections from many levels through the tissue in order to find the site of perforation. The aggregating cell type in perforating diseases also varies. Older lesions tend to have lymphocytes, macrophages or multinucleated giant cells at the perforation site in the dermis. Other lesions may reveal neutrophilic aggregates in the dermis. The Verhoeff-van Gieson (VVG) stain is particularly useful in histological identification of perforating diseases; the stain gives a black appearance to elastic fibers and a red appearance to collagen fibers. (See Figure 1.) Identification of the specific perforating substance is key to differentiating the various perforating disorders. In RPC, there is transepidermal elimination of collagen fibers, whereas the perforating substance in EPS is elastic fibers. Biopsy findings in acquired perforating dermatosis are particularly variable. The histologic finding of transepidermal elimination of both collagen and elastic fibers in patients with hyperkeratotic papules and a history of renal disease or diabetes mellitus was important in the acceptance of acquired perforating dermatosis as a distinct disease entity amongst other perforating disorders.1 Histologic findings in APD can also be identical to perforating folliculitis, childhood EPS, RPC or may be very non-specific with amorphous degenerated material within perforations.6 When APD presents with perforating folliculitis, the perforation may be in the infundibular portion of the hair follicle.18,19 In larger lesions, the perforations may be at the base of the follicular invagination.20,21 MANAGEMENT As pruritus is the most common symptom of APD, antihistamines and other antipruritic agents do provide limited relief. Narrowband UVB phototherapy is the most effective treatment option for patients with APD and renal disease, as it relieves coexisting pruritus.22 Topical, intralesional or systemic corticosteroids or retinoid therapy have been associated with clinical clearance or significant improvement of lesions.2 There are also anecdotal reports of successful treatment with topical menthol, salicylic acid, sulfur and benzoyl peroxide;7 systemic antibiotics such as doxycycline and clindamycin;23,24 and allopurinol in patients with elevated serum uric acid levels.25 Renal transplantation has been associated with cure in occasional patients with dialysis-associated APD.11 Our Patient Our patient’s biopsy was consistent with perforating folliculitis. There was hyperkeratosis and parakeratosis, and a neutrophilic infiltrate within, and perforating the follicular epithelium. (See Figure 2.) The VVG stain showed amorphous degenerated material within the follicle, which is not clearly identifiable as collagen or elastic fibers. Our patient was treated with antihistamines, topical corticosteroids and narrowband UVB phototherapy and noted minimal relief of pruritus. On his last visit in June 2009, he opted to discontinue NBUVB, as new lesions were still occurring on the abdomen and legs. CONCLUSION Acquired perforating dermatosis is a distinct entity within the scope of perforating diseases. It presents with keratotic follicular or perifollicular papules, most frequently over extensor surfaces of the legs. It is important to note that histologically, a lesion may resemble any of the other perforating disorders; or may simply consist of transelimination of necrotic material. A spectrum of different lesions may also be found on a single patient. Therefore, this is a largely clinical diagnosis; and in an adult patient with underlying renal disease or diabetes mellitus, APD should be strongly considered. The exact etiology of APD is unknown, and continuing research may formally identify fibronectin or possibly another pruritogenic factor as the culprit. Early recognition of the signs, symptoms and disease associations in APD can facilitate rapid diagnosis and initiation of appropriate treatment. Dr. Rodney is a dermatology resident in the Department of Dermatology, Howard University Hospital and Veterans Affairs Medical Center, Washington DC. Charla S. Taylor is a medical student at Howard University College of Medicine, Washington DC. Dr. Cohen is former Chief, Department of Dermatology, Veterans Affair Medical Center, Washington DC. Dr. Khachemoune, the Section Editor of Derm Dx, is with Department of Dermatology, State University of New York, Brooklyn, NY. Disclosure: The authors have no conflict of interest with any material presented in this column.