

Puzzling Erythematous Patches Managed with Anakinra
Introduction
Adult-onset Still’s disease (AOSD) is a multisystemic disorder hypothesized to be due to either an autoinflammatory or autoimmune etiology in genetically susceptible patients, similar to the pathogenesis of other conditions such as psoriasis. Generally thought to be due in part to widespread activation of macrophages and neutrophils, pertinent laboratory findings include a discernable leukocytosis with marked neutrophilia, which can aid in the diagnosis. While there remains more to uncover about AOSD, a high-spiking fever, evanescent rash, arthralgias, and pharyngitis are considered clinically characteristic. The advent of Yamaguchi criteria, of which at least five symptoms must be present, has defined required clinical findings to definitively diagnose AOSD. More recently, hyperferritinemia has become a crucial characteristic finding, as it is a marker for macrophage activation and is seen in nearly 70% of cases of AOSD.
Case Report
A previously healthy 37-year-old Mediterranean woman presented to the emergency department for pharyngitis, rhinorrhea, cough, and worsening dyspnea. She was afebrile and had no dermatologic findings upon admission. Two days later, the patient developed acute respiratory failure requiring intubation and mechanical ventilation. Further investigation revealed hepatosplenomegaly, multilobar pulmonary infiltrates, and a pericardial effusion with early tamponade. A pericardial window was placed, with fluid and tissue analysis identifying hemorrhagic and fibrinoid pericarditis with abundant acute and chronic inflammation. The patient developed undulating fevers up to 104.2 °F and was subsequently placed on broad-spectrum antibiotics, which did not improve her condition over the course of 5 days. Extensive viral panel, blood cultures, and sputum cultures were unremarkable; initial laboratory evaluation identified a leukocytosis
(>12,000 cells/mL) with greater than 80% neutrophils.
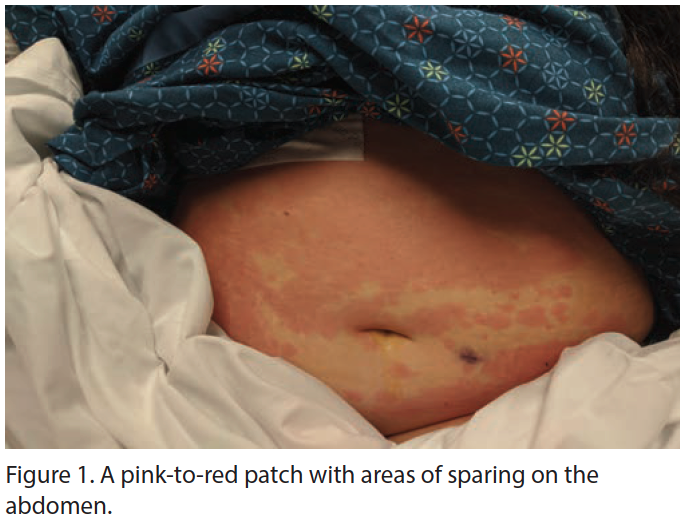
The patient’s antibiotics were narrowed, and she was started on intravenous methylprednisolone (60 mg) and colchicine (1 mg) with dramatic improvement in her symptoms. Extubation was possible after 2 days of this regimen, but her cyclic fevers persisted, generally initiating in the afternoon and lasting 7 to 10 hours before remitting; acetaminophen was ineffective. Dermatology was consulted when evanescent, erythematous patches developed on the patient’s abdomen and lower chest (Figure 1). The patient reported the rash was asymptomatic but appeared to migrate throughout the day; she otherwise denied any previous rashes, arthralgias, abdominal pains, neurologic dysfunction, persistent fevers, or recent travel. Two biopsies were obtained, and extensive autoimmune and hematologic workups were performed.
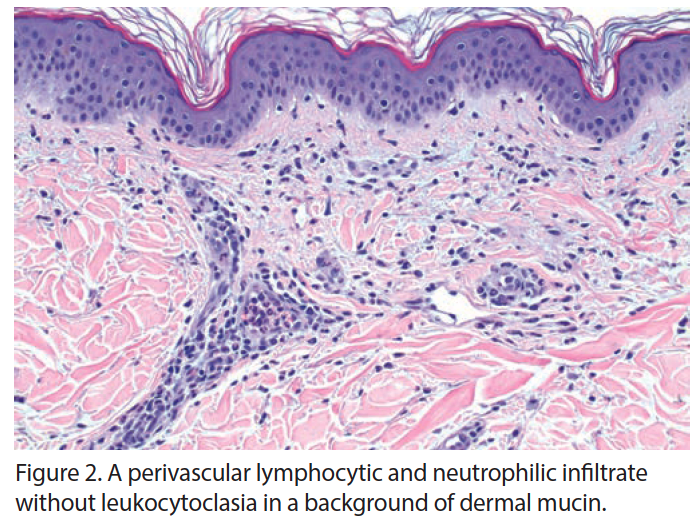
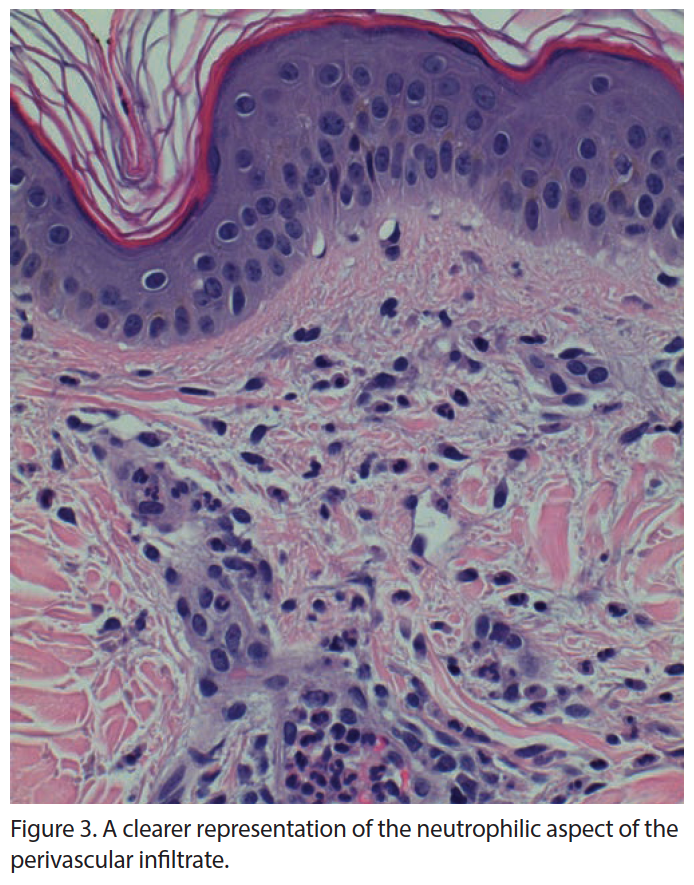
Complete autoimmune panel and serum and urine electrophoresis were unremarkable. Serum ferritin levels were greater than 10,000, the upper limit of the laboratory assay; erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) were increased along with an elevation in liver function tests (LFTs). Dermatopathologic evaluation revealed a perivascular infiltrate of neutrophils with few lymphocytes in the superficial dermis, with no epidermal necrosis, interface change, vasculitis, thrombosis, or leukocytoclasia (Figures 2 and 3). These findings were consistent with an inflammatory pattern of internal origin. Clinicopathologic correlation conferred a working diagnosis of AOSD.
The patient remained stable, but her fevers and ferritin value (>10,000) remained at recheck 1 week after initial evaluation. Rheumatology, whom had been consulted to aid in management, commented that the patient’s condition was consistent with AOSD, now meeting seven of the Yamaguchi criteria. We added 100 mg of anakinra daily to 60 mg daily methylprednisolone with remarkable improvement of the patient’s symptoms. At 4-month follow-up, methylprednisolone has been decreased to 10 mg daily and anakinra has been continued at 100 mg daily; the patient remains completely asymptomatic and all laboratory values have normalized.
Discussion
George Still first coined the term Still’s disease in 1897, after describing 22 children afflicted with what is now called juvenile idiopathic arthritis (JIA).1 AOSD was first described by Eric Bywaters in 1971, after he identified 14 adults suffering from a confluence of symptoms very similar to those seen in JIA.2
Today, AOSD remains a rare systemic autoinflammatory disease of unknown etiology, often difficult to diagnose given its variable presentation and array of diagnostic alternatives.2 Typically affecting young female adults, AOSD may cause life-threatening complications, the most severe of which is macrophage activation syndrome (MAS). Instigated by the presence of abundant proinflammatory cytokines, MAS leads to hypersecretion of interferon (IFN) γ from primed natural killer (NK).2
AOSD is described as a multigenic disorder with characteristics of both autoinflammatory and autoimmune diseases. It is widely postulated that unidentified factors serve as a second hit in those who are genetically susceptible, inciting a pathologic process which culminates in aberrant activation of the inflammatory cascade.1 This marked inflammatory response has been linked to hyperactivity of inflammasomes such as NACHT, LRR, and NLRP3.3 These proteins may respond to so-called “danger signals” of viral or bacterial origin, themselves activating caspases, which overproduce interleukin (IL) 1β.3 Components of both innate and adaptive immune systems have been implicated in the erroneous production of proinflammatory cytokines IL-1β, IL-6, IL-17, IL-18, IFN-γ, and tumor necrosis factor (TNF).1
While familial trends have yet to be reported in AOSD, an association between certain HLA antigens, namely HLA-B17, HLA-B18, HLA-Bw35, and HLA-DR2, and frequency of the disease has been established.4 Of these antigens, HLA-B35 and HLA-DR2 are most strongly associated but do not correlate to disease persistence or severity. Absence of HLA-B35 has been linked to persistent disease, leading some to speculate that HLA-B35 may play a role in driving disease remission.
The clinical presentation of AOSD is protean. The most common manifestations include cyclic high fevers, arthralgias, evanescent rash, and sore throat or pharyngitis.5 Pharyngitis is often the initial presenting symptom and arthralgias are generally reported in the small joints of the distal extremities, but large joints may also be affected, particularly in the acute phase of disease. The evanescent rash is predominantly located on the trunk or proximal extremities and is a salmon-pink color, macular or maculopapular in nature, and commonly appears or becomes more prominent with the onset of fever.5 Serosal involvement is seen in 25% to 60% of patients, with 53% to 82% of patients developing pulmonary symptoms including pleuritis, atelectasis, or diffuse alveolar hemorrhage.4,5 Pouchot et al4 examined 62 patients with AOSD, 37% of whom developed sterile pericarditis; concomitant pleural effusion was identified in 82% of cases. Pertinent laboratory findings include marked leukocytosis, neutrophilia, elevated ESR and CRP, elevated LFTs, and negative autoimmune markers, namely antinuclear antibody (ANA) and rheumatoid factor (RF). Importantly, while the cause remains unknown, hyperferritinemia, an indicator of macrophage activation, seems to be characteristic of AOSD as it is seen in almost 70% of cases.5 Common histologic findings of Still’s rash include necrotic keratinocytes in the upper epidermis and infiltration of lymphocytes and neutrophils in the papillary and middermis.6 This combination allows for histologic
differentiation from most other lichenoid and interface dermatitides and may facilitate earlier diagnosis of AOSD.
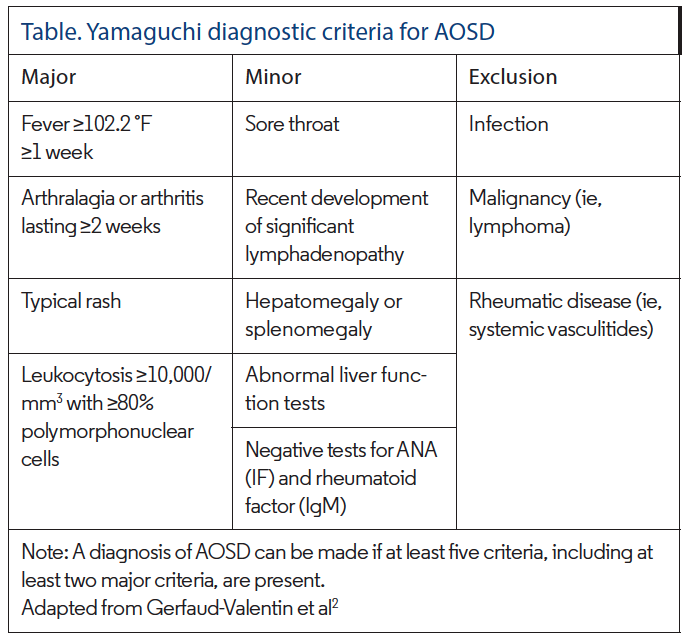
In response to the variable clinical findings in AOSD, six diagnostic scoring systems have been proposed. Yamaguchi criteria are considered most sensitive and have been adopted by most fields, with four major and five minor criteria (Table).7 When five criteria are met, two of which must be major, diagnostic sensitivity is reported to be greater than 96% with specificity of greater than 92%.7
There exists no standard therapeutic ladder for AOSD and treatment remains largely empirical.8 Regimens include nonsteroidal anti-inflammatory drugs (NSAIDs), corticosteroids, and disease-modifying antirheumatic drugs (DMARDs), including biologics and immunologics. Since the efficacy of NSAID monotherapy is poor, with response rates of 20% to 25%, oral corticosteroids, with or without a DMARD (namely, methotrexate), have become the mainstay of therapy; this regimen is effective in approximately 80% of cases.8 Of note, the DMARD sulfasalazine should be used with great caution when treating AOSD, because it may potentiate the onset of MAS, which can be life-threatening; for this reason, it is often bypassed for safer alternatives.9 In the event conventional immunosuppressive therapy is not sufficient, biologic agents targeting IL-1, IL-6, and TNF have shown efficacy.8 Anti-TNF agents, notably infliximab, are often reserved for refractory cases and are generally well tolerated. Infliximab has been associated with adverse events, including infection, fulminant hepatitis, and exacerbation of heart failure, which is particularly ominous in a patient with coexisting pericarditis.8 Some of these complications can be ameliorated or avoided with use of another TNF inhibitory such as etanercept or adalimumab.
Since IL-1β seems to play a dominant role in AOSD, inhibition of IL-1 via anakinra has jumped to the forefront of many treatment algorithms in persistent disease.8 While its effects on
articular symptoms have been less-frequently reported compared with anti-TNF or anti-IL-6 agents, anakinra has been shown to be rapidly efficacious, producing resolution of both systemic and articular manifestations with subsequent normalization of inflammatory markers.8 While limited data has been released thus far, IL-6R inhibitors such as tocilizumab have shown promising results for patients with significant articular and systemic involvement.8 Recent research has identified rituximab as a therapy for patients who have failed all previous treatments, through CD20 targeting and a subsequent decrease in circulating B cells. A report written by Lee and Yoo10 demonstrated resolution of systemic symptoms in a recalcitrant case. Rituximab is also thought to be of value in AOSD patients with concomitant or subsequent thrombotic microangiopathies.10
Conclusions
While rare, AOSD is a serious, multisystemic inflammatory reaction, typically occurring in genetically susceptible young adults. This exaggerated immune response has been linked to hyperactivity of inflammasomes such as NACHT, LRR, and NLRP3, with resultant overproduction of IL-1β. As of yet, there exists no standard therapeutic ladder for AOSD and treatment remains largely empirical. Conventional regimens include NSAIDs, oral corticosteroids, DMARDs, and IL-1 inhibitors.
While more research is needed to illuminate the etiology of AOSD, this review aims to help dermatologists maintain a high index of suspicion when approaching a complicated clinical picture as outlined in this case. As further contributions are made to the literature, proper diagnosis of AOSD will correlate to a prompt initiation of treatment, which will improve patient morbidity and mortality.
Affiliations
Dr White is a fourth-year dermatology resident at HCA Healthcare/USF Morsani College of Medicine: Largo Medical Center, Largo, FL. Mr Brazen is a medical student at Nova Southeastern University: Kiran Patel College of Osteopathic Medicine, Davie, FL. Dr Witfill is an assistant clinical professor of dermatology at HCA Healthcare/USF Morsani College of Medicine: Largo Medical Center.
References
1. Giacomelli R, Ruscitti P, Shoenfeld Y. A comprehensive review on adult onset Still’s disease. J Autoimmun. 2018;93:24-36. doi:10.1016/j.jaut.2018.07.018
2. Gerfaud-Valentin M, Jamilloux Y, Iwaz J, Sève P. Adult-onset Still’s disease. Autoimmun Rev. 2014;13(7):708-722. doi:10.1016/j.autrev.2014.01.058
3. Feist E, Mitrovic S, Fautrel B. Mechanisms, biomarkers and targets for adult-onset Still’s disease. Nat Rev Rheumatol. 2018;14(10):603-618. doi:10.1038/s41584-018-0081-x
4. Pouchot J, Sampalis JS, Beaudet F, et al. Adult Still’s disease: manifestations, disease course, and outcome in 62 patients. Medicine (Baltimore). 1991;70(2):118-136.
5. Kadavath S, Efthimiou P. Adult-onset Still’s disease-pathogenesis, clinical manifestations, and new treatment options. Ann Med. 2015;47(1):6-14. doi:10.3109/07853890.2014.971052
6. Lee JY, Yang CC, Hsu MM. Histopathology of persistent papules and plaques in adult-onset Still’s disease. J Am Acad Dermatol. 2005;52(6):1003-1008.
doi:10.1016/j.jaad.2005.02.032
7. Yamaguchi M, Ohta A, Tsunematsu T, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992;19(3):424-430.
8. Sfriso P, Priori R, Valesini G, et al. Adult-onset Still’s disease: an Italian multicentre retrospective observational study of manifestations and treatments in 245 patients. Clin Rheumatol. 2016;35(7):1683-1689. doi:10.1007/s10067-016-3308-8
9. Yoo DH. Treatment of adult-onset still’s disease: up to date. Expert Rev Clin Immunol. 2017;13(9):849-866. doi:10.1080/1744666X.2017.1332994
10. Lee WS, Yoo WH. Rituximab for refractory adult-onset Still’s disease with thrombotic microangiopathy. Rheumatology (Oxford). 2014;53(9):1717-1718. doi:10.1093/rheumatology/keu027

Current Issue


Subscribe










