A 48-year-old man presented with this growth on the nose in June 2014 but he declined biopsy to confirm our clinical diagnosis (Figure 1). He was informed about risks associated with no treatment. He returned at the end of August 2015 (Figure 2).

What is your diagnosis?
Answer on page 2
{{pagebreak}}
Diagnosis:
Regressive Keratoacanthoma
Keratoacanthomas (KAs) are relatively common epidermal tumors characterized by rapid growth and subsequent regression. Sharing many clinical and histologic similarities with cutaneous squamous cell carcinomas (cSCC), these tumors have represented an area of controversy in the field of dermatology since the 1950s.1 Whether KAs represent highly differentiated cSCC or a unique category of dermatologic pseudo-malignancy has not been entirely elucidated and remains an area of active research. Due to the close clinical and histopathologic resemblance between cSCC and KA, it is not yet possible to predict which lesions may spontaneously regress and which have the potential to become metastatic. As such, many clinicians choose to treat potential KAs as cSCC to minimize patient morbidity and mortality.
What Is a KA?
KAs are tumors derived from the pilosebaceous glands of the skin and are characterized by rapid growth and self-resolution over weeks to months. Due to their striking resemblance to SCC, debate exists as to whether they simply represent a subtype of cSCC (ie, true malignancy) or rather, a distinct and benign entity that has a unique ability to self-resolve. Though the etiology of KA is not entirely known, UV light appears to play an integral role in their development.2 As such, they are typically observed in elderly individuals with fair skin and a background of other sunlight-induced skin diseases.3 The link between UV light and the development of KA is further strengthened by the increase in incidence seen in the summer and fall months.4 Other risk factors that have been associated with KA formation include trauma,5 immunosuppression,6 chemical carcinogens,7 and human papillomavirus infection.8
An epidemiological study conducted in Australia indicated that the annual incidence of KAs in fair-skinned individuals is as high as 150 per 100,000.9 However, the incidence in individuals with darker skin types is far less with ethnic Japanese, Filipino, and Hawaiian populations being reported as 22, 7, and 6 per 100,000, respectively.10 It should be noted that the true incidence may be underreported as lesions that undergo regression may go unrecognized by physicians.1 For comparative purposes, the incidence ratio of KA to cSCC ranges between 1:0.6 and 1:5, with differences depending on geographic location.10
Clinical Presentation
KAs are classically described as sharply demarcated dome-shaped, skin-colored nodules with central keratinous plugs that are most commonly found as solitary lesions on the face, neck, and dorsal hands (Figure 1). Their distinct crateriform appearance along with their rapid growth helps to distinguish these lesions from other cutaneous diseases such as nonmelanoma skin cancer. Within 6 to 8 weeks, KAs can grow to a diameter of 1.0 to 2.5 cm and, in doing so, may cause local tissue destruction.11 A KA typically begins as a pink macule that progresses to a papule before finally assuming its keratin-filled volcano-like appearance. Once full growth is achieved, up to 50% can be expected to resolve spontaneously over another 4 to 6 weeks until only an atrophic or hypopigmented scar remains (Figure 2).12 However, this progression may not always be observed. Reports of more aggressive KA have been described whereby the lesion demonstrates metastatic potential and also transformation into cSCC.13 This atypical tumor behavior is more commonly seen in immunocompromised and elderly individuals.14

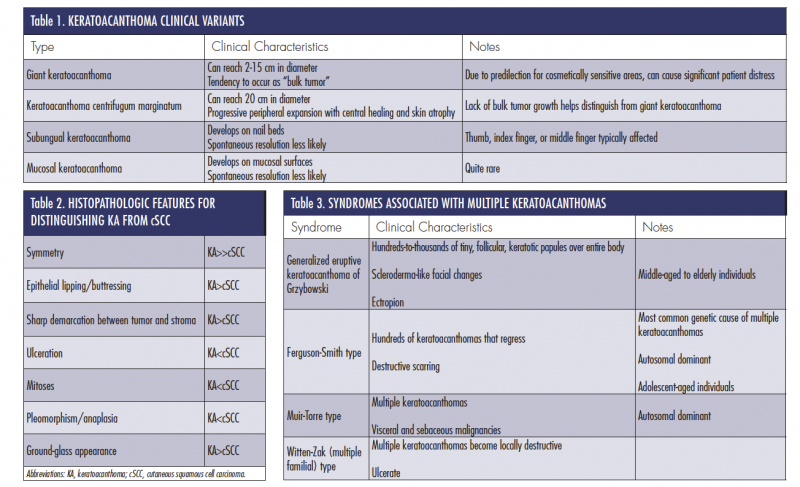
Though the aforementioned clinical description and progression is classic for most solitary KAs, 4 clinically distinct subtypes have been described in the literature (Table 1).15-18
Histologic Presentation
Histologically, KA is distinguished by central invagination with a characteristic hyperkeratotic core and “lipping” (ie, “buttressing”) of the epidermis over the peripheral rim of the central keratotic plug. Epidermal hyperplasia is present, but nuclear atypia and mitoses are not prominent features. Additionally, there is typically a sharp demarcation between the tumor and surrounding stroma as well as a mixed inflammatory infiltrate within the dermis.19
There is a significant amount of overlap between the histologic features of KA and cSCC. This has led to cases of misdiagnosis with subsequent mismanagement of the 2 lesions. Kwiek and Schwartz19 recently identified 7 of the most useful histopathologic features for KA and cSCC differentiation (Table 2).
Associated Syndromes
Several syndromes are associated with the development of multiple KAs. The following 4 are described in Table 3: generalized eruptive KA of Grzybowski, Ferguson-Smith type, Muir-Torre type, and Witten-Zak (multiple familial) type.17-22
Metastatic KA and Controversies
The most significant remaining controversy surrounding KA can be summarized in the following question: Does KA carry true malignant and metastatic potential? Metastatic behavior is generally considered unlikely, yet several case reports have been published describing such behavior.23,24 Predicting the behavior of KA based on histologic presentation alone can be difficult. Researchers have suggested that perineural invasion, which occurs in 1% to 4% of KAs, may be a risk factor for aggressive behavior.25 However, larger studies have identified a low frequency of aggressive behavior in such cases.26 Thus, at the present, perineural invasion is currently considered an incidental finding with no impact on prognosis. A second view regarding the metastatic potential of KA is that the few cases of “metastatic KA” that have been documented were likely a misdiagnosis, and that a diagnosis of cSCC probably would have been more accurate had clearer histopathologic guidelines for distinguishing the 2 existed at the time of presentation.27
A second related controversy that has persisted for decades is the following: are these lesions simply a subtype of cSCC or are they truly a distinct and benign entity with the unique ability to self-resolve. The evaluation of biologic markers has helped to shed some light on this issue. An evaluation of 17 moderately differentiated cSCC compared with 24 early-proliferative phase KAs yielded the following results: stronger expression of p53 mutations (71% vs 8%), telomerase activity (65% vs 17%), and cyclooxygenase-2 expression (65% vs 8%) in cSCC vs KAs.28 While it is impractical to use these markers in routine clinical practice, these findings are nevertheless important in identifying the 2 as distinct entities. Furthermore, an understanding of their unique genetic expressions helps to explain the different clinical behavior of the 2 lesions.
Article continues on page 3
{{pagebreak}}
Observation vs Active Treatment
It is generally accepted as best practice to actively treat KAs as opposed to serial observation for spontaneous resolution, though spontaneous resolution is the most likely natural history for the lesion. The rationale for this approach is based on 2 factors: the first is that cSCC develops in an average of 6% of KAs. This incidence increases in direct relation to the age of the patient, reaching 14% in patients older than 90 years.29 Thus, the most prudent method for decreasing occurrence of cSCC associated with KA is removal of the tumor. The second part of the rationale for active treatment of KA is that many patients experience embarrassment by the appearance of the lesions. Simple excision, or other forms of removal, will help to prevent any social ramifications for patients.
Management and Pitfalls
First-line therapy for patients presenting with a KA is surgical excision. The procedure is simple, well tolerated, and provides histopathologic confirmation of tumor removal. Several other acceptable therapies also have been reported in the literature. These include electrodesiccation and curettage, intralesional therapy with 5-fluorouracil or methotrexate, topical application of 5-fluorouracil or imiquimod, or radiation therapy.
Additionally, patients should be given the option to defer therapy. Guidelines for patients pursuing this course of action are listed in Table 4.

Generalized eruptive KA of Grzybowski requires special consideration as it relates to management, as simple excision is not practical for the hundreds-to-thousands of lesions that a patient may present with. In this case, oral retinoids such as isotretinoin, acitretin, and etretinate have shown good efficacy.30 Patients should be informed that complete resolution may require trials with several different dosages and that complete resolution may take several years.
Our Patient
After the patient was informed of the diagnosis, treatment options (including deferring treatment) were discussed in detail. He decided to forego all active treatments and opted for observation only. Over the next 3 months, spontaneous regression led to the complete resolution of the KA. The patient will be following up with the dermatologist regularly.
Conclusion
KAs are epidermal tumors that arise from the pilosebaceous glands of the skin. These skin lesions are characterized by a rapid growth phase followed by spontaneous resolution in most cases. Alternate growth and regression patterns have been associated with patients who have an immunocompromised status as well as those who are elderly. These populations are more likely to experience atypical tumor behavior with the possibility of metastasis and malignant transformation. A variety of genetic syndromes associated with multiple KAs have been described in the literature, though they remain rare causes of KA formation. Future research is still needed to further elucidate the precise pathogenesis of these unique dermatologic tumors, as a definitive consensus regarding their etiology is still a topic of debate among dermatologists.
Mr Caravaglio is with the University of Central Florida College of Medicine in Orlando, FL.
Mr Kelly is with University of Central Florida College of Medicine in Orlando, FL.
Dr Mamouni is in private practice in Oum El Bouaghi, Algeria.
 Dr Khachemoune, the Section Editor of Derm DX, is with the department of dermatology at Veteran Affairs Medical Center, and the department of dermatology at the State University of New York Downstate, both in Brooklyn, NY.
Dr Khachemoune, the Section Editor of Derm DX, is with the department of dermatology at Veteran Affairs Medical Center, and the department of dermatology at the State University of New York Downstate, both in Brooklyn, NY.
Disclosure: The authors report no relevant financial relationships.
References
1. Karaa A, Khachemoune A. Keratoacanthoma: a tumor in search of a classification. Int J Dermatol. 2007;46(7):671-678.
2. Craddock KJ, Rao J, Lauzon GJ, Tron VA. Multiple keratoacanthomas arising post-UVB therapy. J Cutan Med Surg. 2004;8(4):239-243.
3. Chowdhury RK, Padhi T, Das GS. Keratoacanthoma of the conjunctiva complicating xeroderma pigmentosum. Indian J Dermatol Venereol Leprol. 2005;71(6):430-431.
4. Dufresne RG, Marrero GM, Robinson-Bostom L. Seasonal presentation of keratoacanthomas in Rhode Island. Br J Dermatol. 1997;136(2):227-229.
5. Pattee SF, Silvis NG. Keratoacanthoma developing in sites of previous trauma: a report of two cases and review of the literature. J Am Acad Dermatol. 2003;48(2 suppl):S35-S38.
6. Poleksic S, Yeung KY. Rapid development of keratoacanthoma and accelerated transformation into squamous cell carcinoma of the skin: a mutagenic effect of polychemotherapy in a patient with Hodgkin’s disease? Cancer. 1978;41(1):12-16.
7. Heidenreich RK, Gongloff RK, Wescott WB. A solitary, exophytic, crateriform lesion on the mandibular retromolar gingiva. J Am Dent Assoc. 1986;112(3):377-379.
8. Stockfleth E, Meinke B, Arndt R, Christophers E, Meyer T. Identification of DNA sequences of both genital and cutaneous HPV types in a small number of keratoacanthomas of nonimmunosuppressed patients. Dermatology. 1999;198(2):122-125.
9. Sullivan JJ. Keratoacanthoma: the Australian experience. Australas J Dermatol. 1997;38(suppl 1):S36-S39.
10. Chuang TY, Reizner GT, Elpern DJ, Stone JL, Farmer ER. Non-melanoma skin cancer and keratoacanthoma in Filipinos: an incidence report from Kauai, Hawaii. Int J Dermatol. 1993;32(10):717-718.
11. Kwittken J. A histologic chronology of the clinical course of the keratocarcinoma (so-called keratoacanthoma). Mt Sinai J Med.1975;42(2):127-135.
12. Seifert A, Nasemann T. Keratoacanthoma and its clinical variants. Review of the literature and histopathologic analysis of 90 cases. Hautarzt. 1989;40(4):189-202.
13. Sánchez Yus E, Simón P, Requena L, Ambrojo P, de Eusebio E. Solitary keratoacanthoma: a self-healing proliferation that frequently becomes malignant. Am J Dermatopathol. 2000;22(4):305-310.
14. Hodak E, Jones RE, Ackerman AB. Solitary keratoacanthoma is a squamous-cell carcinoma: three examples with metastases. Am J Dermatopathol. 1993;15(4):332-342.
15. Hofer SO, Jackson IT. Self-involution of giant keratoacanthoma on the tip of the nose. Plast Reconstr Surg. 2004;113(2):765-766.
16. Sung CS, Chuang FC, Ho JC, Lin SH. Keratoacanthoma centrifugum marginatum—A rare variant of keratoacanthoma: Case report and literature review. Dermatologica Sinica. 2014;32(1):25-28.
17. Schwartz RA. Keratoacanthoma. J Am Acad Dermatol. 1994;30(1):1-19; quiz 20-22.
18. Borkhatariya PB, Gupta S, Bang D, Rawal RC. Keratoacanthoma centrifugum marginatum: case report and review of literature. Indian J Dermatol. 2011;56(4):455-456.
19. Kwiek B, Schwartz RA. Keratoacanthoma (KA): An update and review. J Am Acad Dermatol. 2016;74(6):1220-1233.
20. Smith JF. Multiple primary, self-healing squamous epithelioma of the skin. Br J Dermatol. 1948;60(10):315-318.
21. Ponti G, Losi L, Di Gregorio C, et al. Identification of Muir-Torre syndrome among patients with sebaceous tumors and keratoacanthomas: role of clinical features, microsatellite instability, and immunohistochemistry. Cancer. 2005;103(5):1018-1025.
22. Agarwal M, Chander R, Karmakar S, Walia R. Multiple familial keratoacanthoma of Witten and Zak–a report of three siblings. Dermatology. 1999;198(4):396-399.
23. Ansai S, Manabe M. Possible spontaneous regression of a metastatic lesion of keratoacanthoma-like squamous cell carcinoma in a regional lymph node. J Dermatol. 2005;32(11):899-903.
24. Davis BA, Monheit GD, Kline L. Metastatic skin cancer presenting as ptosis and diplopia. Dermatol Surg. 2006;32(1):148-158.
25. Patrie M, Yehuda E, Carmen C. Keratoacanthoma of the head and neck with perineural invasion: incidental finding or cause for concern? Dermatol Surg. 2010;36(7):1209-1213.
26. Godbolt AM, Sullivan JJ, Weedon D. Keratoacanthoma with perineural invasion: a report of 40 cases. Australas J Dermatol. 2001;42(3):168-171.
27. Mandrell JC, Santa Cruz D. Keratoacanthoma: hyperplasia, benign neoplasm, or a type of squamous cell carcinoma? Semin Diagn Pathol. 2009;26(3):150-163.
28. Putti TC, Teh M, Lee YS. Biological behavior of keratoacanthoma and squamous cell carcinoma: telomerase activity and COX-2 as potential markers. Mod Pathol. 2004;17(4):468-475.
29. Weedon DD, Malo J, Brooks D, Williamson R. Squamous cell carcinoma arising in keratoacanthoma: a neglected phenomenon in the elderly. Am J Dermatopathol. 2010;32(5):423-426.
30. Vandergriff T, Nakamura K, High WA. Generalized eruptive keratoacanthomas of Grzybowski treated with isotretinoin. J Drugs Dermatol. 2008;7(11):1069-1071.
A 48-year-old man presented with this growth on the nose in June 2014 but he declined biopsy to confirm our clinical diagnosis (Figure 1). He was informed about risks associated with no treatment. He returned at the end of August 2015 (Figure 2).

What is your diagnosis?
Diagnosis:
Regressive Keratoacanthoma
Keratoacanthomas (KAs) are relatively common epidermal tumors characterized by rapid growth and subsequent regression. Sharing many clinical and histologic similarities with cutaneous squamous cell carcinomas (cSCC), these tumors have represented an area of controversy in the field of dermatology since the 1950s.1 Whether KAs represent highly differentiated cSCC or a unique category of dermatologic pseudo-malignancy has not been entirely elucidated and remains an area of active research. Due to the close clinical and histopathologic resemblance between cSCC and KA, it is not yet possible to predict which lesions may spontaneously regress and which have the potential to become metastatic. As such, many clinicians choose to treat potential KAs as cSCC to minimize patient morbidity and mortality.
What Is a KA?
KAs are tumors derived from the pilosebaceous glands of the skin and are characterized by rapid growth and self-resolution over weeks to months. Due to their striking resemblance to SCC, debate exists as to whether they simply represent a subtype of cSCC (ie, true malignancy) or rather, a distinct and benign entity that has a unique ability to self-resolve. Though the etiology of KA is not entirely known, UV light appears to play an integral role in their development.2 As such, they are typically observed in elderly individuals with fair skin and a background of other sunlight-induced skin diseases.3 The link between UV light and the development of KA is further strengthened by the increase in incidence seen in the summer and fall months.4 Other risk factors that have been associated with KA formation include trauma,5 immunosuppression,6 chemical carcinogens,7 and human papillomavirus infection.8
An epidemiological study conducted in Australia indicated that the annual incidence of KAs in fair-skinned individuals is as high as 150 per 100,000.9 However, the incidence in individuals with darker skin types is far less with ethnic Japanese, Filipino, and Hawaiian populations being reported as 22, 7, and 6 per 100,000, respectively.10 It should be noted that the true incidence may be underreported as lesions that undergo regression may go unrecognized by physicians.1 For comparative purposes, the incidence ratio of KA to cSCC ranges between 1:0.6 and 1:5, with differences depending on geographic location.10
Clinical Presentation
KAs are classically described as sharply demarcated dome-shaped, skin-colored nodules with central keratinous plugs that are most commonly found as solitary lesions on the face, neck, and dorsal hands (Figure 1). Their distinct crateriform appearance along with their rapid growth helps to distinguish these lesions from other cutaneous diseases such as nonmelanoma skin cancer. Within 6 to 8 weeks, KAs can grow to a diameter of 1.0 to 2.5 cm and, in doing so, may cause local tissue destruction.11 A KA typically begins as a pink macule that progresses to a papule before finally assuming its keratin-filled volcano-like appearance. Once full growth is achieved, up to 50% can be expected to resolve spontaneously over another 4 to 6 weeks until only an atrophic or hypopigmented scar remains (Figure 2).12 However, this progression may not always be observed. Reports of more aggressive KA have been described whereby the lesion demonstrates metastatic potential and also transformation into cSCC.13 This atypical tumor behavior is more commonly seen in immunocompromised and elderly individuals.14

Though the aforementioned clinical description and progression is classic for most solitary KAs, 4 clinically distinct subtypes have been described in the literature (Table 1).15-18
Histologic Presentation
Histologically, KA is distinguished by central invagination with a characteristic hyperkeratotic core and “lipping” (ie, “buttressing”) of the epidermis over the peripheral rim of the central keratotic plug. Epidermal hyperplasia is present, but nuclear atypia and mitoses are not prominent features. Additionally, there is typically a sharp demarcation between the tumor and surrounding stroma as well as a mixed inflammatory infiltrate within the dermis.19
There is a significant amount of overlap between the histologic features of KA and cSCC. This has led to cases of misdiagnosis with subsequent mismanagement of the 2 lesions. Kwiek and Schwartz19 recently identified 7 of the most useful histopathologic features for KA and cSCC differentiation (Table 2).
Associated Syndromes
Several syndromes are associated with the development of multiple KAs. The following 4 are described in Table 3: generalized eruptive KA of Grzybowski, Ferguson-Smith type, Muir-Torre type, and Witten-Zak (multiple familial) type.17-22

Metastatic KA and Controversies
The most significant remaining controversy surrounding KA can be summarized in the following question: Does KA carry true malignant and metastatic potential? Metastatic behavior is generally considered unlikely, yet several case reports have been published describing such behavior.23,24 Predicting the behavior of KA based on histologic presentation alone can be difficult. Researchers have suggested that perineural invasion, which occurs in 1% to 4% of KAs, may be a risk factor for aggressive behavior.25 However, larger studies have identified a low frequency of aggressive behavior in such cases.26 Thus, at the present, perineural invasion is currently considered an incidental finding with no impact on prognosis. A second view regarding the metastatic potential of KA is that the few cases of “metastatic KA” that have been documented were likely a misdiagnosis, and that a diagnosis of cSCC probably would have been more accurate had clearer histopathologic guidelines for distinguishing the 2 existed at the time of presentation.27
A second related controversy that has persisted for decades is the following: are these lesions simply a subtype of cSCC or are they truly a distinct and benign entity with the unique ability to self-resolve. The evaluation of biologic markers has helped to shed some light on this issue. An evaluation of 17 moderately differentiated cSCC compared with 24 early-proliferative phase KAs yielded the following results: stronger expression of p53 mutations (71% vs 8%), telomerase activity (65% vs 17%), and cyclooxygenase-2 expression (65% vs 8%) in cSCC vs KAs.28 While it is impractical to use these markers in routine clinical practice, these findings are nevertheless important in identifying the 2 as distinct entities. Furthermore, an understanding of their unique genetic expressions helps to explain the different clinical behavior of the 2 lesions.
Article continues on page 3
{{pagebreak}}
Observation vs Active Treatment
It is generally accepted as best practice to actively treat KAs as opposed to serial observation for spontaneous resolution, though spontaneous resolution is the most likely natural history for the lesion. The rationale for this approach is based on 2 factors: the first is that cSCC develops in an average of 6% of KAs. This incidence increases in direct relation to the age of the patient, reaching 14% in patients older than 90 years.29 Thus, the most prudent method for decreasing occurrence of cSCC associated with KA is removal of the tumor. The second part of the rationale for active treatment of KA is that many patients experience embarrassment by the appearance of the lesions. Simple excision, or other forms of removal, will help to prevent any social ramifications for patients.
Management and Pitfalls
First-line therapy for patients presenting with a KA is surgical excision. The procedure is simple, well tolerated, and provides histopathologic confirmation of tumor removal. Several other acceptable therapies also have been reported in the literature. These include electrodesiccation and curettage, intralesional therapy with 5-fluorouracil or methotrexate, topical application of 5-fluorouracil or imiquimod, or radiation therapy.
Additionally, patients should be given the option to defer therapy. Guidelines for patients pursuing this course of action are listed in Table 4.

Generalized eruptive KA of Grzybowski requires special consideration as it relates to management, as simple excision is not practical for the hundreds-to-thousands of lesions that a patient may present with. In this case, oral retinoids such as isotretinoin, acitretin, and etretinate have shown good efficacy.30 Patients should be informed that complete resolution may require trials with several different dosages and that complete resolution may take several years.
Our Patient
After the patient was informed of the diagnosis, treatment options (including deferring treatment) were discussed in detail. He decided to forego all active treatments and opted for observation only. Over the next 3 months, spontaneous regression led to the complete resolution of the KA. The patient will be following up with the dermatologist regularly.
Conclusion
KAs are epidermal tumors that arise from the pilosebaceous glands of the skin. These skin lesions are characterized by a rapid growth phase followed by spontaneous resolution in most cases. Alternate growth and regression patterns have been associated with patients who have an immunocompromised status as well as those who are elderly. These populations are more likely to experience atypical tumor behavior with the possibility of metastasis and malignant transformation. A variety of genetic syndromes associated with multiple KAs have been described in the literature, though they remain rare causes of KA formation. Future research is still needed to further elucidate the precise pathogenesis of these unique dermatologic tumors, as a definitive consensus regarding their etiology is still a topic of debate among dermatologists.
Mr Caravaglio is with the University of Central Florida College of Medicine in Orlando, FL.
Mr Kelly is with University of Central Florida College of Medicine in Orlando, FL.
Dr Mamouni is in private practice in Oum El Bouaghi, Algeria.
 Dr Khachemoune, the Section Editor of Derm DX, is with the department of dermatology at Veteran Affairs Medical Center, and the department of dermatology at the State University of New York Downstate, both in Brooklyn, NY.
Dr Khachemoune, the Section Editor of Derm DX, is with the department of dermatology at Veteran Affairs Medical Center, and the department of dermatology at the State University of New York Downstate, both in Brooklyn, NY.
Disclosure: The authors report no relevant financial relationships.
References
1. Karaa A, Khachemoune A. Keratoacanthoma: a tumor in search of a classification. Int J Dermatol. 2007;46(7):671-678.
2. Craddock KJ, Rao J, Lauzon GJ, Tron VA. Multiple keratoacanthomas arising post-UVB therapy. J Cutan Med Surg. 2004;8(4):239-243.
3. Chowdhury RK, Padhi T, Das GS. Keratoacanthoma of the conjunctiva complicating xeroderma pigmentosum. Indian J Dermatol Venereol Leprol. 2005;71(6):430-431.
4. Dufresne RG, Marrero GM, Robinson-Bostom L. Seasonal presentation of keratoacanthomas in Rhode Island. Br J Dermatol. 1997;136(2):227-229.
5. Pattee SF, Silvis NG. Keratoacanthoma developing in sites of previous trauma: a report of two cases and review of the literature. J Am Acad Dermatol. 2003;48(2 suppl):S35-S38.
6. Poleksic S, Yeung KY. Rapid development of keratoacanthoma and accelerated transformation into squamous cell carcinoma of the skin: a mutagenic effect of polychemotherapy in a patient with Hodgkin’s disease? Cancer. 1978;41(1):12-16.
7. Heidenreich RK, Gongloff RK, Wescott WB. A solitary, exophytic, crateriform lesion on the mandibular retromolar gingiva. J Am Dent Assoc. 1986;112(3):377-379.
8. Stockfleth E, Meinke B, Arndt R, Christophers E, Meyer T. Identification of DNA sequences of both genital and cutaneous HPV types in a small number of keratoacanthomas of nonimmunosuppressed patients. Dermatology. 1999;198(2):122-125.
9. Sullivan JJ. Keratoacanthoma: the Australian experience. Australas J Dermatol. 1997;38(suppl 1):S36-S39.
10. Chuang TY, Reizner GT, Elpern DJ, Stone JL, Farmer ER. Non-melanoma skin cancer and keratoacanthoma in Filipinos: an incidence report from Kauai, Hawaii. Int J Dermatol. 1993;32(10):717-718.
11. Kwittken J. A histologic chronology of the clinical course of the keratocarcinoma (so-called keratoacanthoma). Mt Sinai J Med.1975;42(2):127-135.
12. Seifert A, Nasemann T. Keratoacanthoma and its clinical variants. Review of the literature and histopathologic analysis of 90 cases. Hautarzt. 1989;40(4):189-202.
13. Sánchez Yus E, Simón P, Requena L, Ambrojo P, de Eusebio E. Solitary keratoacanthoma: a self-healing proliferation that frequently becomes malignant. Am J Dermatopathol. 2000;22(4):305-310.
14. Hodak E, Jones RE, Ackerman AB. Solitary keratoacanthoma is a squamous-cell carcinoma: three examples with metastases. Am J Dermatopathol. 1993;15(4):332-342.
15. Hofer SO, Jackson IT. Self-involution of giant keratoacanthoma on the tip of the nose. Plast Reconstr Surg. 2004;113(2):765-766.
16. Sung CS, Chuang FC, Ho JC, Lin SH. Keratoacanthoma centrifugum marginatum—A rare variant of keratoacanthoma: Case report and literature review. Dermatologica Sinica. 2014;32(1):25-28.
17. Schwartz RA. Keratoacanthoma. J Am Acad Dermatol. 1994;30(1):1-19; quiz 20-22.
18. Borkhatariya PB, Gupta S, Bang D, Rawal RC. Keratoacanthoma centrifugum marginatum: case report and review of literature. Indian J Dermatol. 2011;56(4):455-456.
19. Kwiek B, Schwartz RA. Keratoacanthoma (KA): An update and review. J Am Acad Dermatol. 2016;74(6):1220-1233.
20. Smith JF. Multiple primary, self-healing squamous epithelioma of the skin. Br J Dermatol. 1948;60(10):315-318.
21. Ponti G, Losi L, Di Gregorio C, et al. Identification of Muir-Torre syndrome among patients with sebaceous tumors and keratoacanthomas: role of clinical features, microsatellite instability, and immunohistochemistry. Cancer. 2005;103(5):1018-1025.
22. Agarwal M, Chander R, Karmakar S, Walia R. Multiple familial keratoacanthoma of Witten and Zak–a report of three siblings. Dermatology. 1999;198(4):396-399.
23. Ansai S, Manabe M. Possible spontaneous regression of a metastatic lesion of keratoacanthoma-like squamous cell carcinoma in a regional lymph node. J Dermatol. 2005;32(11):899-903.
24. Davis BA, Monheit GD, Kline L. Metastatic skin cancer presenting as ptosis and diplopia. Dermatol Surg. 2006;32(1):148-158.
25. Patrie M, Yehuda E, Carmen C. Keratoacanthoma of the head and neck with perineural invasion: incidental finding or cause for concern? Dermatol Surg. 2010;36(7):1209-1213.
26. Godbolt AM, Sullivan JJ, Weedon D. Keratoacanthoma with perineural invasion: a report of 40 cases. Australas J Dermatol. 2001;42(3):168-171.
27. Mandrell JC, Santa Cruz D. Keratoacanthoma: hyperplasia, benign neoplasm, or a type of squamous cell carcinoma? Semin Diagn Pathol. 2009;26(3):150-163.
28. Putti TC, Teh M, Lee YS. Biological behavior of keratoacanthoma and squamous cell carcinoma: telomerase activity and COX-2 as potential markers. Mod Pathol. 2004;17(4):468-475.
29. Weedon DD, Malo J, Brooks D, Williamson R. Squamous cell carcinoma arising in keratoacanthoma: a neglected phenomenon in the elderly. Am J Dermatopathol. 2010;32(5):423-426.
30. Vandergriff T, Nakamura K, High WA. Generalized eruptive keratoacanthomas of Grzybowski treated with isotretinoin. J Drugs Dermatol. 2008;7(11):1069-1071.
A 48-year-old man presented with this growth on the nose in June 2014 but he declined biopsy to confirm our clinical diagnosis (Figure 1). He was informed about risks associated with no treatment. He returned at the end of August 2015 (Figure 2).
What is your diagnosis?
,
A 48-year-old man presented with this growth on the nose in June 2014 but he declined biopsy to confirm our clinical diagnosis (Figure 1). He was informed about risks associated with no treatment. He returned at the end of August 2015 (Figure 2).
What is your diagnosis?
Answer on page 2
{{pagebreak}}
Diagnosis:
Regressive Keratoacanthoma
Keratoacanthomas (KAs) are relatively common epidermal tumors characterized by rapid growth and subsequent regression. Sharing many clinical and histologic similarities with cutaneous squamous cell carcinomas (cSCC), these tumors have represented an area of controversy in the field of dermatology since the 1950s.1 Whether KAs represent highly differentiated cSCC or a unique category of dermatologic pseudo-malignancy has not been entirely elucidated and remains an area of active research. Due to the close clinical and histopathologic resemblance between cSCC and KA, it is not yet possible to predict which lesions may spontaneously regress and which have the potential to become metastatic. As such, many clinicians choose to treat potential KAs as cSCC to minimize patient morbidity and mortality.
What Is a KA?
KAs are tumors derived from the pilosebaceous glands of the skin and are characterized by rapid growth and self-resolution over weeks to months. Due to their striking resemblance to SCC, debate exists as to whether they simply represent a subtype of cSCC (ie, true malignancy) or rather, a distinct and benign entity that has a unique ability to self-resolve. Though the etiology of KA is not entirely known, UV light appears to play an integral role in their development.2 As such, they are typically observed in elderly individuals with fair skin and a background of other sunlight-induced skin diseases.3 The link between UV light and the development of KA is further strengthened by the increase in incidence seen in the summer and fall months.4 Other risk factors that have been associated with KA formation include trauma,5 immunosuppression,6 chemical carcinogens,7 and human papillomavirus infection.8
An epidemiological study conducted in Australia indicated that the annual incidence of KAs in fair-skinned individuals is as high as 150 per 100,000.9 However, the incidence in individuals with darker skin types is far less with ethnic Japanese, Filipino, and Hawaiian populations being reported as 22, 7, and 6 per 100,000, respectively.10 It should be noted that the true incidence may be underreported as lesions that undergo regression may go unrecognized by physicians.1 For comparative purposes, the incidence ratio of KA to cSCC ranges between 1:0.6 and 1:5, with differences depending on geographic location.10
Clinical Presentation
KAs are classically described as sharply demarcated dome-shaped, skin-colored nodules with central keratinous plugs that are most commonly found as solitary lesions on the face, neck, and dorsal hands (Figure 1). Their distinct crateriform appearance along with their rapid growth helps to distinguish these lesions from other cutaneous diseases such as nonmelanoma skin cancer. Within 6 to 8 weeks, KAs can grow to a diameter of 1.0 to 2.5 cm and, in doing so, may cause local tissue destruction.11 A KA typically begins as a pink macule that progresses to a papule before finally assuming its keratin-filled volcano-like appearance. Once full growth is achieved, up to 50% can be expected to resolve spontaneously over another 4 to 6 weeks until only an atrophic or hypopigmented scar remains (Figure 2).12 However, this progression may not always be observed. Reports of more aggressive KA have been described whereby the lesion demonstrates metastatic potential and also transformation into cSCC.13 This atypical tumor behavior is more commonly seen in immunocompromised and elderly individuals.14
Though the aforementioned clinical description and progression is classic for most solitary KAs, 4 clinically distinct subtypes have been described in the literature (Table 1).15-18
Histologic Presentation
Histologically, KA is distinguished by central invagination with a characteristic hyperkeratotic core and “lipping” (ie, “buttressing”) of the epidermis over the peripheral rim of the central keratotic plug. Epidermal hyperplasia is present, but nuclear atypia and mitoses are not prominent features. Additionally, there is typically a sharp demarcation between the tumor and surrounding stroma as well as a mixed inflammatory infiltrate within the dermis.19
There is a significant amount of overlap between the histologic features of KA and cSCC. This has led to cases of misdiagnosis with subsequent mismanagement of the 2 lesions. Kwiek and Schwartz19 recently identified 7 of the most useful histopathologic features for KA and cSCC differentiation (Table 2).
Associated Syndromes
Several syndromes are associated with the development of multiple KAs. The following 4 are described in Table 3: generalized eruptive KA of Grzybowski, Ferguson-Smith type, Muir-Torre type, and Witten-Zak (multiple familial) type.17-22
Metastatic KA and Controversies
The most significant remaining controversy surrounding KA can be summarized in the following question: Does KA carry true malignant and metastatic potential? Metastatic behavior is generally considered unlikely, yet several case reports have been published describing such behavior.23,24 Predicting the behavior of KA based on histologic presentation alone can be difficult. Researchers have suggested that perineural invasion, which occurs in 1% to 4% of KAs, may be a risk factor for aggressive behavior.25 However, larger studies have identified a low frequency of aggressive behavior in such cases.26 Thus, at the present, perineural invasion is currently considered an incidental finding with no impact on prognosis. A second view regarding the metastatic potential of KA is that the few cases of “metastatic KA” that have been documented were likely a misdiagnosis, and that a diagnosis of cSCC probably would have been more accurate had clearer histopathologic guidelines for distinguishing the 2 existed at the time of presentation.27
A second related controversy that has persisted for decades is the following: are these lesions simply a subtype of cSCC or are they truly a distinct and benign entity with the unique ability to self-resolve. The evaluation of biologic markers has helped to shed some light on this issue. An evaluation of 17 moderately differentiated cSCC compared with 24 early-proliferative phase KAs yielded the following results: stronger expression of p53 mutations (71% vs 8%), telomerase activity (65% vs 17%), and cyclooxygenase-2 expression (65% vs 8%) in cSCC vs KAs.28 While it is impractical to use these markers in routine clinical practice, these findings are nevertheless important in identifying the 2 as distinct entities. Furthermore, an understanding of their unique genetic expressions helps to explain the different clinical behavior of the 2 lesions.
Article continues on page 3
{{pagebreak}}
Observation vs Active Treatment
It is generally accepted as best practice to actively treat KAs as opposed to serial observation for spontaneous resolution, though spontaneous resolution is the most likely natural history for the lesion. The rationale for this approach is based on 2 factors: the first is that cSCC develops in an average of 6% of KAs. This incidence increases in direct relation to the age of the patient, reaching 14% in patients older than 90 years.29 Thus, the most prudent method for decreasing occurrence of cSCC associated with KA is removal of the tumor. The second part of the rationale for active treatment of KA is that many patients experience embarrassment by the appearance of the lesions. Simple excision, or other forms of removal, will help to prevent any social ramifications for patients.
Management and Pitfalls
First-line therapy for patients presenting with a KA is surgical excision. The procedure is simple, well tolerated, and provides histopathologic confirmation of tumor removal. Several other acceptable therapies also have been reported in the literature. These include electrodesiccation and curettage, intralesional therapy with 5-fluorouracil or methotrexate, topical application of 5-fluorouracil or imiquimod, or radiation therapy.
Additionally, patients should be given the option to defer therapy. Guidelines for patients pursuing this course of action are listed in Table 4.
Generalized eruptive KA of Grzybowski requires special consideration as it relates to management, as simple excision is not practical for the hundreds-to-thousands of lesions that a patient may present with. In this case, oral retinoids such as isotretinoin, acitretin, and etretinate have shown good efficacy.30 Patients should be informed that complete resolution may require trials with several different dosages and that complete resolution may take several years.
Our Patient
After the patient was informed of the diagnosis, treatment options (including deferring treatment) were discussed in detail. He decided to forego all active treatments and opted for observation only. Over the next 3 months, spontaneous regression led to the complete resolution of the KA. The patient will be following up with the dermatologist regularly.
Conclusion
KAs are epidermal tumors that arise from the pilosebaceous glands of the skin. These skin lesions are characterized by a rapid growth phase followed by spontaneous resolution in most cases. Alternate growth and regression patterns have been associated with patients who have an immunocompromised status as well as those who are elderly. These populations are more likely to experience atypical tumor behavior with the possibility of metastasis and malignant transformation. A variety of genetic syndromes associated with multiple KAs have been described in the literature, though they remain rare causes of KA formation. Future research is still needed to further elucidate the precise pathogenesis of these unique dermatologic tumors, as a definitive consensus regarding their etiology is still a topic of debate among dermatologists.
Mr Caravaglio is with the University of Central Florida College of Medicine in Orlando, FL.
Mr Kelly is with University of Central Florida College of Medicine in Orlando, FL.
Dr Mamouni is in private practice in Oum El Bouaghi, Algeria.
Dr Khachemoune, the Section Editor of Derm DX, is with the department of dermatology at Veteran Affairs Medical Center, and the department of dermatology at the State University of New York Downstate, both in Brooklyn, NY.
Disclosure: The authors report no relevant financial relationships.
References
1. Karaa A, Khachemoune A. Keratoacanthoma: a tumor in search of a classification. Int J Dermatol. 2007;46(7):671-678.
2. Craddock KJ, Rao J, Lauzon GJ, Tron VA. Multiple keratoacanthomas arising post-UVB therapy. J Cutan Med Surg. 2004;8(4):239-243.
3. Chowdhury RK, Padhi T, Das GS. Keratoacanthoma of the conjunctiva complicating xeroderma pigmentosum. Indian J Dermatol Venereol Leprol. 2005;71(6):430-431.
4. Dufresne RG, Marrero GM, Robinson-Bostom L. Seasonal presentation of keratoacanthomas in Rhode Island. Br J Dermatol. 1997;136(2):227-229.
5. Pattee SF, Silvis NG. Keratoacanthoma developing in sites of previous trauma: a report of two cases and review of the literature. J Am Acad Dermatol. 2003;48(2 suppl):S35-S38.
6. Poleksic S, Yeung KY. Rapid development of keratoacanthoma and accelerated transformation into squamous cell carcinoma of the skin: a mutagenic effect of polychemotherapy in a patient with Hodgkin’s disease? Cancer. 1978;41(1):12-16.
7. Heidenreich RK, Gongloff RK, Wescott WB. A solitary, exophytic, crateriform lesion on the mandibular retromolar gingiva. J Am Dent Assoc. 1986;112(3):377-379.
8. Stockfleth E, Meinke B, Arndt R, Christophers E, Meyer T. Identification of DNA sequences of both genital and cutaneous HPV types in a small number of keratoacanthomas of nonimmunosuppressed patients. Dermatology. 1999;198(2):122-125.
9. Sullivan JJ. Keratoacanthoma: the Australian experience. Australas J Dermatol. 1997;38(suppl 1):S36-S39.
10. Chuang TY, Reizner GT, Elpern DJ, Stone JL, Farmer ER. Non-melanoma skin cancer and keratoacanthoma in Filipinos: an incidence report from Kauai, Hawaii. Int J Dermatol. 1993;32(10):717-718.
11. Kwittken J. A histologic chronology of the clinical course of the keratocarcinoma (so-called keratoacanthoma). Mt Sinai J Med.1975;42(2):127-135.
12. Seifert A, Nasemann T. Keratoacanthoma and its clinical variants. Review of the literature and histopathologic analysis of 90 cases. Hautarzt. 1989;40(4):189-202.
13. Sánchez Yus E, Simón P, Requena L, Ambrojo P, de Eusebio E. Solitary keratoacanthoma: a self-healing proliferation that frequently becomes malignant. Am J Dermatopathol. 2000;22(4):305-310.
14. Hodak E, Jones RE, Ackerman AB. Solitary keratoacanthoma is a squamous-cell carcinoma: three examples with metastases. Am J Dermatopathol. 1993;15(4):332-342.
15. Hofer SO, Jackson IT. Self-involution of giant keratoacanthoma on the tip of the nose. Plast Reconstr Surg. 2004;113(2):765-766.
16. Sung CS, Chuang FC, Ho JC, Lin SH. Keratoacanthoma centrifugum marginatum—A rare variant of keratoacanthoma: Case report and literature review. Dermatologica Sinica. 2014;32(1):25-28.
17. Schwartz RA. Keratoacanthoma. J Am Acad Dermatol. 1994;30(1):1-19; quiz 20-22.
18. Borkhatariya PB, Gupta S, Bang D, Rawal RC. Keratoacanthoma centrifugum marginatum: case report and review of literature. Indian J Dermatol. 2011;56(4):455-456.
19. Kwiek B, Schwartz RA. Keratoacanthoma (KA): An update and review. J Am Acad Dermatol. 2016;74(6):1220-1233.
20. Smith JF. Multiple primary, self-healing squamous epithelioma of the skin. Br J Dermatol. 1948;60(10):315-318.
21. Ponti G, Losi L, Di Gregorio C, et al. Identification of Muir-Torre syndrome among patients with sebaceous tumors and keratoacanthomas: role of clinical features, microsatellite instability, and immunohistochemistry. Cancer. 2005;103(5):1018-1025.
22. Agarwal M, Chander R, Karmakar S, Walia R. Multiple familial keratoacanthoma of Witten and Zak–a report of three siblings. Dermatology. 1999;198(4):396-399.
23. Ansai S, Manabe M. Possible spontaneous regression of a metastatic lesion of keratoacanthoma-like squamous cell carcinoma in a regional lymph node. J Dermatol. 2005;32(11):899-903.
24. Davis BA, Monheit GD, Kline L. Metastatic skin cancer presenting as ptosis and diplopia. Dermatol Surg. 2006;32(1):148-158.
25. Patrie M, Yehuda E, Carmen C. Keratoacanthoma of the head and neck with perineural invasion: incidental finding or cause for concern? Dermatol Surg. 2010;36(7):1209-1213.
26. Godbolt AM, Sullivan JJ, Weedon D. Keratoacanthoma with perineural invasion: a report of 40 cases. Australas J Dermatol. 2001;42(3):168-171.
27. Mandrell JC, Santa Cruz D. Keratoacanthoma: hyperplasia, benign neoplasm, or a type of squamous cell carcinoma? Semin Diagn Pathol. 2009;26(3):150-163.
28. Putti TC, Teh M, Lee YS. Biological behavior of keratoacanthoma and squamous cell carcinoma: telomerase activity and COX-2 as potential markers. Mod Pathol. 2004;17(4):468-475.
29. Weedon DD, Malo J, Brooks D, Williamson R. Squamous cell carcinoma arising in keratoacanthoma: a neglected phenomenon in the elderly. Am J Dermatopathol. 2010;32(5):423-426.
30. Vandergriff T, Nakamura K, High WA. Generalized eruptive keratoacanthomas of Grzybowski treated with isotretinoin. J Drugs Dermatol. 2008;7(11):1069-1071.
A 48-year-old man presented with this growth on the nose in June 2014 but he declined biopsy to confirm our clinical diagnosis (Figure 1). He was informed about risks associated with no treatment. He returned at the end of August 2015 (Figure 2).
What is your diagnosis?
Diagnosis:
Regressive Keratoacanthoma
Keratoacanthomas (KAs) are relatively common epidermal tumors characterized by rapid growth and subsequent regression. Sharing many clinical and histologic similarities with cutaneous squamous cell carcinomas (cSCC), these tumors have represented an area of controversy in the field of dermatology since the 1950s.1 Whether KAs represent highly differentiated cSCC or a unique category of dermatologic pseudo-malignancy has not been entirely elucidated and remains an area of active research. Due to the close clinical and histopathologic resemblance between cSCC and KA, it is not yet possible to predict which lesions may spontaneously regress and which have the potential to become metastatic. As such, many clinicians choose to treat potential KAs as cSCC to minimize patient morbidity and mortality.
What Is a KA?
KAs are tumors derived from the pilosebaceous glands of the skin and are characterized by rapid growth and self-resolution over weeks to months. Due to their striking resemblance to SCC, debate exists as to whether they simply represent a subtype of cSCC (ie, true malignancy) or rather, a distinct and benign entity that has a unique ability to self-resolve. Though the etiology of KA is not entirely known, UV light appears to play an integral role in their development.2 As such, they are typically observed in elderly individuals with fair skin and a background of other sunlight-induced skin diseases.3 The link between UV light and the development of KA is further strengthened by the increase in incidence seen in the summer and fall months.4 Other risk factors that have been associated with KA formation include trauma,5 immunosuppression,6 chemical carcinogens,7 and human papillomavirus infection.8
An epidemiological study conducted in Australia indicated that the annual incidence of KAs in fair-skinned individuals is as high as 150 per 100,000.9 However, the incidence in individuals with darker skin types is far less with ethnic Japanese, Filipino, and Hawaiian populations being reported as 22, 7, and 6 per 100,000, respectively.10 It should be noted that the true incidence may be underreported as lesions that undergo regression may go unrecognized by physicians.1 For comparative purposes, the incidence ratio of KA to cSCC ranges between 1:0.6 and 1:5, with differences depending on geographic location.10
Clinical Presentation
KAs are classically described as sharply demarcated dome-shaped, skin-colored nodules with central keratinous plugs that are most commonly found as solitary lesions on the face, neck, and dorsal hands (Figure 1). Their distinct crateriform appearance along with their rapid growth helps to distinguish these lesions from other cutaneous diseases such as nonmelanoma skin cancer. Within 6 to 8 weeks, KAs can grow to a diameter of 1.0 to 2.5 cm and, in doing so, may cause local tissue destruction.11 A KA typically begins as a pink macule that progresses to a papule before finally assuming its keratin-filled volcano-like appearance. Once full growth is achieved, up to 50% can be expected to resolve spontaneously over another 4 to 6 weeks until only an atrophic or hypopigmented scar remains (Figure 2).12 However, this progression may not always be observed. Reports of more aggressive KA have been described whereby the lesion demonstrates metastatic potential and also transformation into cSCC.13 This atypical tumor behavior is more commonly seen in immunocompromised and elderly individuals.14
Though the aforementioned clinical description and progression is classic for most solitary KAs, 4 clinically distinct subtypes have been described in the literature (Table 1).15-18
Histologic Presentation
Histologically, KA is distinguished by central invagination with a characteristic hyperkeratotic core and “lipping” (ie, “buttressing”) of the epidermis over the peripheral rim of the central keratotic plug. Epidermal hyperplasia is present, but nuclear atypia and mitoses are not prominent features. Additionally, there is typically a sharp demarcation between the tumor and surrounding stroma as well as a mixed inflammatory infiltrate within the dermis.19
There is a significant amount of overlap between the histologic features of KA and cSCC. This has led to cases of misdiagnosis with subsequent mismanagement of the 2 lesions. Kwiek and Schwartz19 recently identified 7 of the most useful histopathologic features for KA and cSCC differentiation (Table 2).
Associated Syndromes
Several syndromes are associated with the development of multiple KAs. The following 4 are described in Table 3: generalized eruptive KA of Grzybowski, Ferguson-Smith type, Muir-Torre type, and Witten-Zak (multiple familial) type.17-22
Metastatic KA and Controversies
The most significant remaining controversy surrounding KA can be summarized in the following question: Does KA carry true malignant and metastatic potential? Metastatic behavior is generally considered unlikely, yet several case reports have been published describing such behavior.23,24 Predicting the behavior of KA based on histologic presentation alone can be difficult. Researchers have suggested that perineural invasion, which occurs in 1% to 4% of KAs, may be a risk factor for aggressive behavior.25 However, larger studies have identified a low frequency of aggressive behavior in such cases.26 Thus, at the present, perineural invasion is currently considered an incidental finding with no impact on prognosis. A second view regarding the metastatic potential of KA is that the few cases of “metastatic KA” that have been documented were likely a misdiagnosis, and that a diagnosis of cSCC probably would have been more accurate had clearer histopathologic guidelines for distinguishing the 2 existed at the time of presentation.27
A second related controversy that has persisted for decades is the following: are these lesions simply a subtype of cSCC or are they truly a distinct and benign entity with the unique ability to self-resolve. The evaluation of biologic markers has helped to shed some light on this issue. An evaluation of 17 moderately differentiated cSCC compared with 24 early-proliferative phase KAs yielded the following results: stronger expression of p53 mutations (71% vs 8%), telomerase activity (65% vs 17%), and cyclooxygenase-2 expression (65% vs 8%) in cSCC vs KAs.28 While it is impractical to use these markers in routine clinical practice, these findings are nevertheless important in identifying the 2 as distinct entities. Furthermore, an understanding of their unique genetic expressions helps to explain the different clinical behavior of the 2 lesions.
Article continues on page 3
{{pagebreak}}
Observation vs Active Treatment
It is generally accepted as best practice to actively treat KAs as opposed to serial observation for spontaneous resolution, though spontaneous resolution is the most likely natural history for the lesion. The rationale for this approach is based on 2 factors: the first is that cSCC develops in an average of 6% of KAs. This incidence increases in direct relation to the age of the patient, reaching 14% in patients older than 90 years.29 Thus, the most prudent method for decreasing occurrence of cSCC associated with KA is removal of the tumor. The second part of the rationale for active treatment of KA is that many patients experience embarrassment by the appearance of the lesions. Simple excision, or other forms of removal, will help to prevent any social ramifications for patients.
Management and Pitfalls
First-line therapy for patients presenting with a KA is surgical excision. The procedure is simple, well tolerated, and provides histopathologic confirmation of tumor removal. Several other acceptable therapies also have been reported in the literature. These include electrodesiccation and curettage, intralesional therapy with 5-fluorouracil or methotrexate, topical application of 5-fluorouracil or imiquimod, or radiation therapy.
Additionally, patients should be given the option to defer therapy. Guidelines for patients pursuing this course of action are listed in Table 4.
Generalized eruptive KA of Grzybowski requires special consideration as it relates to management, as simple excision is not practical for the hundreds-to-thousands of lesions that a patient may present with. In this case, oral retinoids such as isotretinoin, acitretin, and etretinate have shown good efficacy.30 Patients should be informed that complete resolution may require trials with several different dosages and that complete resolution may take several years.
Our Patient
After the patient was informed of the diagnosis, treatment options (including deferring treatment) were discussed in detail. He decided to forego all active treatments and opted for observation only. Over the next 3 months, spontaneous regression led to the complete resolution of the KA. The patient will be following up with the dermatologist regularly.
Conclusion
KAs are epidermal tumors that arise from the pilosebaceous glands of the skin. These skin lesions are characterized by a rapid growth phase followed by spontaneous resolution in most cases. Alternate growth and regression patterns have been associated with patients who have an immunocompromised status as well as those who are elderly. These populations are more likely to experience atypical tumor behavior with the possibility of metastasis and malignant transformation. A variety of genetic syndromes associated with multiple KAs have been described in the literature, though they remain rare causes of KA formation. Future research is still needed to further elucidate the precise pathogenesis of these unique dermatologic tumors, as a definitive consensus regarding their etiology is still a topic of debate among dermatologists.
Mr Caravaglio is with the University of Central Florida College of Medicine in Orlando, FL.
Mr Kelly is with University of Central Florida College of Medicine in Orlando, FL.
Dr Mamouni is in private practice in Oum El Bouaghi, Algeria.
Dr Khachemoune, the Section Editor of Derm DX, is with the department of dermatology at Veteran Affairs Medical Center, and the department of dermatology at the State University of New York Downstate, both in Brooklyn, NY.
Disclosure: The authors report no relevant financial relationships.
References
1. Karaa A, Khachemoune A. Keratoacanthoma: a tumor in search of a classification. Int J Dermatol. 2007;46(7):671-678.
2. Craddock KJ, Rao J, Lauzon GJ, Tron VA. Multiple keratoacanthomas arising post-UVB therapy. J Cutan Med Surg. 2004;8(4):239-243.
3. Chowdhury RK, Padhi T, Das GS. Keratoacanthoma of the conjunctiva complicating xeroderma pigmentosum. Indian J Dermatol Venereol Leprol. 2005;71(6):430-431.
4. Dufresne RG, Marrero GM, Robinson-Bostom L. Seasonal presentation of keratoacanthomas in Rhode Island. Br J Dermatol. 1997;136(2):227-229.
5. Pattee SF, Silvis NG. Keratoacanthoma developing in sites of previous trauma: a report of two cases and review of the literature. J Am Acad Dermatol. 2003;48(2 suppl):S35-S38.
6. Poleksic S, Yeung KY. Rapid development of keratoacanthoma and accelerated transformation into squamous cell carcinoma of the skin: a mutagenic effect of polychemotherapy in a patient with Hodgkin’s disease? Cancer. 1978;41(1):12-16.
7. Heidenreich RK, Gongloff RK, Wescott WB. A solitary, exophytic, crateriform lesion on the mandibular retromolar gingiva. J Am Dent Assoc. 1986;112(3):377-379.
8. Stockfleth E, Meinke B, Arndt R, Christophers E, Meyer T. Identification of DNA sequences of both genital and cutaneous HPV types in a small number of keratoacanthomas of nonimmunosuppressed patients. Dermatology. 1999;198(2):122-125.
9. Sullivan JJ. Keratoacanthoma: the Australian experience. Australas J Dermatol. 1997;38(suppl 1):S36-S39.
10. Chuang TY, Reizner GT, Elpern DJ, Stone JL, Farmer ER. Non-melanoma skin cancer and keratoacanthoma in Filipinos: an incidence report from Kauai, Hawaii. Int J Dermatol. 1993;32(10):717-718.
11. Kwittken J. A histologic chronology of the clinical course of the keratocarcinoma (so-called keratoacanthoma). Mt Sinai J Med.1975;42(2):127-135.
12. Seifert A, Nasemann T. Keratoacanthoma and its clinical variants. Review of the literature and histopathologic analysis of 90 cases. Hautarzt. 1989;40(4):189-202.
13. Sánchez Yus E, Simón P, Requena L, Ambrojo P, de Eusebio E. Solitary keratoacanthoma: a self-healing proliferation that frequently becomes malignant. Am J Dermatopathol. 2000;22(4):305-310.
14. Hodak E, Jones RE, Ackerman AB. Solitary keratoacanthoma is a squamous-cell carcinoma: three examples with metastases. Am J Dermatopathol. 1993;15(4):332-342.
15. Hofer SO, Jackson IT. Self-involution of giant keratoacanthoma on the tip of the nose. Plast Reconstr Surg. 2004;113(2):765-766.
16. Sung CS, Chuang FC, Ho JC, Lin SH. Keratoacanthoma centrifugum marginatum—A rare variant of keratoacanthoma: Case report and literature review. Dermatologica Sinica. 2014;32(1):25-28.
17. Schwartz RA. Keratoacanthoma. J Am Acad Dermatol. 1994;30(1):1-19; quiz 20-22.
18. Borkhatariya PB, Gupta S, Bang D, Rawal RC. Keratoacanthoma centrifugum marginatum: case report and review of literature. Indian J Dermatol. 2011;56(4):455-456.
19. Kwiek B, Schwartz RA. Keratoacanthoma (KA): An update and review. J Am Acad Dermatol. 2016;74(6):1220-1233.
20. Smith JF. Multiple primary, self-healing squamous epithelioma of the skin. Br J Dermatol. 1948;60(10):315-318.
21. Ponti G, Losi L, Di Gregorio C, et al. Identification of Muir-Torre syndrome among patients with sebaceous tumors and keratoacanthomas: role of clinical features, microsatellite instability, and immunohistochemistry. Cancer. 2005;103(5):1018-1025.
22. Agarwal M, Chander R, Karmakar S, Walia R. Multiple familial keratoacanthoma of Witten and Zak–a report of three siblings. Dermatology. 1999;198(4):396-399.
23. Ansai S, Manabe M. Possible spontaneous regression of a metastatic lesion of keratoacanthoma-like squamous cell carcinoma in a regional lymph node. J Dermatol. 2005;32(11):899-903.
24. Davis BA, Monheit GD, Kline L. Metastatic skin cancer presenting as ptosis and diplopia. Dermatol Surg. 2006;32(1):148-158.
25. Patrie M, Yehuda E, Carmen C. Keratoacanthoma of the head and neck with perineural invasion: incidental finding or cause for concern? Dermatol Surg. 2010;36(7):1209-1213.
26. Godbolt AM, Sullivan JJ, Weedon D. Keratoacanthoma with perineural invasion: a report of 40 cases. Australas J Dermatol. 2001;42(3):168-171.
27. Mandrell JC, Santa Cruz D. Keratoacanthoma: hyperplasia, benign neoplasm, or a type of squamous cell carcinoma? Semin Diagn Pathol. 2009;26(3):150-163.
28. Putti TC, Teh M, Lee YS. Biological behavior of keratoacanthoma and squamous cell carcinoma: telomerase activity and COX-2 as potential markers. Mod Pathol. 2004;17(4):468-475.
29. Weedon DD, Malo J, Brooks D, Williamson R. Squamous cell carcinoma arising in keratoacanthoma: a neglected phenomenon in the elderly. Am J Dermatopathol. 2010;32(5):423-426.
30. Vandergriff T, Nakamura K, High WA. Generalized eruptive keratoacanthomas of Grzybowski treated with isotretinoin. J Drugs Dermatol. 2008;7(11):1069-1071.
Diagnosis:
Regressive Keratoacanthoma
Keratoacanthomas (KAs) are relatively common epidermal tumors characterized by rapid growth and subsequent regression. Sharing many clinical and histologic similarities with cutaneous squamous cell carcinomas (cSCC), these tumors have represented an area of controversy in the field of dermatology since the 1950s.1 Whether KAs represent highly differentiated cSCC or a unique category of dermatologic pseudo-malignancy has not been entirely elucidated and remains an area of active research. Due to the close clinical and histopathologic resemblance between cSCC and KA, it is not yet possible to predict which lesions may spontaneously regress and which have the potential to become metastatic. As such, many clinicians choose to treat potential KAs as cSCC to minimize patient morbidity and mortality.
What Is a KA?
KAs are tumors derived from the pilosebaceous glands of the skin and are characterized by rapid growth and self-resolution over weeks to months. Due to their striking resemblance to SCC, debate exists as to whether they simply represent a subtype of cSCC (ie, true malignancy) or rather, a distinct and benign entity that has a unique ability to self-resolve. Though the etiology of KA is not entirely known, UV light appears to play an integral role in their development.2 As such, they are typically observed in elderly individuals with fair skin and a background of other sunlight-induced skin diseases.3 The link between UV light and the development of KA is further strengthened by the increase in incidence seen in the summer and fall months.4 Other risk factors that have been associated with KA formation include trauma,5 immunosuppression,6 chemical carcinogens,7 and human papillomavirus infection.8
An epidemiological study conducted in Australia indicated that the annual incidence of KAs in fair-skinned individuals is as high as 150 per 100,000.9 However, the incidence in individuals with darker skin types is far less with ethnic Japanese, Filipino, and Hawaiian populations being reported as 22, 7, and 6 per 100,000, respectively.10 It should be noted that the true incidence may be underreported as lesions that undergo regression may go unrecognized by physicians.1 For comparative purposes, the incidence ratio of KA to cSCC ranges between 1:0.6 and 1:5, with differences depending on geographic location.10
Clinical Presentation
KAs are classically described as sharply demarcated dome-shaped, skin-colored nodules with central keratinous plugs that are most commonly found as solitary lesions on the face, neck, and dorsal hands (Figure 1). Their distinct crateriform appearance along with their rapid growth helps to distinguish these lesions from other cutaneous diseases such as nonmelanoma skin cancer. Within 6 to 8 weeks, KAs can grow to a diameter of 1.0 to 2.5 cm and, in doing so, may cause local tissue destruction.11 A KA typically begins as a pink macule that progresses to a papule before finally assuming its keratin-filled volcano-like appearance. Once full growth is achieved, up to 50% can be expected to resolve spontaneously over another 4 to 6 weeks until only an atrophic or hypopigmented scar remains (Figure 2).12 However, this progression may not always be observed. Reports of more aggressive KA have been described whereby the lesion demonstrates metastatic potential and also transformation into cSCC.13 This atypical tumor behavior is more commonly seen in immunocompromised and elderly individuals.14
Though the aforementioned clinical description and progression is classic for most solitary KAs, 4 clinically distinct subtypes have been described in the literature (Table 1).15-18
Histologic Presentation
Histologically, KA is distinguished by central invagination with a characteristic hyperkeratotic core and “lipping” (ie, “buttressing”) of the epidermis over the peripheral rim of the central keratotic plug. Epidermal hyperplasia is present, but nuclear atypia and mitoses are not prominent features. Additionally, there is typically a sharp demarcation between the tumor and surrounding stroma as well as a mixed inflammatory infiltrate within the dermis.19
There is a significant amount of overlap between the histologic features of KA and cSCC. This has led to cases of misdiagnosis with subsequent mismanagement of the 2 lesions. Kwiek and Schwartz19 recently identified 7 of the most useful histopathologic features for KA and cSCC differentiation (Table 2).
Associated Syndromes
Several syndromes are associated with the development of multiple KAs. The following 4 are described in Table 3: generalized eruptive KA of Grzybowski, Ferguson-Smith type, Muir-Torre type, and Witten-Zak (multiple familial) type.17-22
Metastatic KA and Controversies
The most significant remaining controversy surrounding KA can be summarized in the following question: Does KA carry true malignant and metastatic potential? Metastatic behavior is generally considered unlikely, yet several case reports have been published describing such behavior.23,24 Predicting the behavior of KA based on histologic presentation alone can be difficult. Researchers have suggested that perineural invasion, which occurs in 1% to 4% of KAs, may be a risk factor for aggressive behavior.25 However, larger studies have identified a low frequency of aggressive behavior in such cases.26 Thus, at the present, perineural invasion is currently considered an incidental finding with no impact on prognosis. A second view regarding the metastatic potential of KA is that the few cases of “metastatic KA” that have been documented were likely a misdiagnosis, and that a diagnosis of cSCC probably would have been more accurate had clearer histopathologic guidelines for distinguishing the 2 existed at the time of presentation.27
A second related controversy that has persisted for decades is the following: are these lesions simply a subtype of cSCC or are they truly a distinct and benign entity with the unique ability to self-resolve. The evaluation of biologic markers has helped to shed some light on this issue. An evaluation of 17 moderately differentiated cSCC compared with 24 early-proliferative phase KAs yielded the following results: stronger expression of p53 mutations (71% vs 8%), telomerase activity (65% vs 17%), and cyclooxygenase-2 expression (65% vs 8%) in cSCC vs KAs.28 While it is impractical to use these markers in routine clinical practice, these findings are nevertheless important in identifying the 2 as distinct entities. Furthermore, an understanding of their unique genetic expressions helps to explain the different clinical behavior of the 2 lesions.
Article continues on page 3
{{pagebreak}}
Observation vs Active Treatment
It is generally accepted as best practice to actively treat KAs as opposed to serial observation for spontaneous resolution, though spontaneous resolution is the most likely natural history for the lesion. The rationale for this approach is based on 2 factors: the first is that cSCC develops in an average of 6% of KAs. This incidence increases in direct relation to the age of the patient, reaching 14% in patients older than 90 years.29 Thus, the most prudent method for decreasing occurrence of cSCC associated with KA is removal of the tumor. The second part of the rationale for active treatment of KA is that many patients experience embarrassment by the appearance of the lesions. Simple excision, or other forms of removal, will help to prevent any social ramifications for patients.
Management and Pitfalls
First-line therapy for patients presenting with a KA is surgical excision. The procedure is simple, well tolerated, and provides histopathologic confirmation of tumor removal. Several other acceptable therapies also have been reported in the literature. These include electrodesiccation and curettage, intralesional therapy with 5-fluorouracil or methotrexate, topical application of 5-fluorouracil or imiquimod, or radiation therapy.
Additionally, patients should be given the option to defer therapy. Guidelines for patients pursuing this course of action are listed in Table 4.
Generalized eruptive KA of Grzybowski requires special consideration as it relates to management, as simple excision is not practical for the hundreds-to-thousands of lesions that a patient may present with. In this case, oral retinoids such as isotretinoin, acitretin, and etretinate have shown good efficacy.30 Patients should be informed that complete resolution may require trials with several different dosages and that complete resolution may take several years.
Our Patient
After the patient was informed of the diagnosis, treatment options (including deferring treatment) were discussed in detail. He decided to forego all active treatments and opted for observation only. Over the next 3 months, spontaneous regression led to the complete resolution of the KA. The patient will be following up with the dermatologist regularly.
Conclusion
KAs are epidermal tumors that arise from the pilosebaceous glands of the skin. These skin lesions are characterized by a rapid growth phase followed by spontaneous resolution in most cases. Alternate growth and regression patterns have been associated with patients who have an immunocompromised status as well as those who are elderly. These populations are more likely to experience atypical tumor behavior with the possibility of metastasis and malignant transformation. A variety of genetic syndromes associated with multiple KAs have been described in the literature, though they remain rare causes of KA formation. Future research is still needed to further elucidate the precise pathogenesis of these unique dermatologic tumors, as a definitive consensus regarding their etiology is still a topic of debate among dermatologists.
Mr Caravaglio is with the University of Central Florida College of Medicine in Orlando, FL.
Mr Kelly is with University of Central Florida College of Medicine in Orlando, FL.
Dr Mamouni is in private practice in Oum El Bouaghi, Algeria.
Dr Khachemoune, the Section Editor of Derm DX, is with the department of dermatology at Veteran Affairs Medical Center, and the department of dermatology at the State University of New York Downstate, both in Brooklyn, NY.
Disclosure: The authors report no relevant financial relationships.
References
1. Karaa A, Khachemoune A. Keratoacanthoma: a tumor in search of a classification. Int J Dermatol. 2007;46(7):671-678.
2. Craddock KJ, Rao J, Lauzon GJ, Tron VA. Multiple keratoacanthomas arising post-UVB therapy. J Cutan Med Surg. 2004;8(4):239-243.
3. Chowdhury RK, Padhi T, Das GS. Keratoacanthoma of the conjunctiva complicating xeroderma pigmentosum. Indian J Dermatol Venereol Leprol. 2005;71(6):430-431.
4. Dufresne RG, Marrero GM, Robinson-Bostom L. Seasonal presentation of keratoacanthomas in Rhode Island. Br J Dermatol. 1997;136(2):227-229.
5. Pattee SF, Silvis NG. Keratoacanthoma developing in sites of previous trauma: a report of two cases and review of the literature. J Am Acad Dermatol. 2003;48(2 suppl):S35-S38.
6. Poleksic S, Yeung KY. Rapid development of keratoacanthoma and accelerated transformation into squamous cell carcinoma of the skin: a mutagenic effect of polychemotherapy in a patient with Hodgkin’s disease? Cancer. 1978;41(1):12-16.
7. Heidenreich RK, Gongloff RK, Wescott WB. A solitary, exophytic, crateriform lesion on the mandibular retromolar gingiva. J Am Dent Assoc. 1986;112(3):377-379.
8. Stockfleth E, Meinke B, Arndt R, Christophers E, Meyer T. Identification of DNA sequences of both genital and cutaneous HPV types in a small number of keratoacanthomas of nonimmunosuppressed patients. Dermatology. 1999;198(2):122-125.
9. Sullivan JJ. Keratoacanthoma: the Australian experience. Australas J Dermatol. 1997;38(suppl 1):S36-S39.
10. Chuang TY, Reizner GT, Elpern DJ, Stone JL, Farmer ER. Non-melanoma skin cancer and keratoacanthoma in Filipinos: an incidence report from Kauai, Hawaii. Int J Dermatol. 1993;32(10):717-718.
11. Kwittken J. A histologic chronology of the clinical course of the keratocarcinoma (so-called keratoacanthoma). Mt Sinai J Med.1975;42(2):127-135.
12. Seifert A, Nasemann T. Keratoacanthoma and its clinical variants. Review of the literature and histopathologic analysis of 90 cases. Hautarzt. 1989;40(4):189-202.
13. Sánchez Yus E, Simón P, Requena L, Ambrojo P, de Eusebio E. Solitary keratoacanthoma: a self-healing proliferation that frequently becomes malignant. Am J Dermatopathol. 2000;22(4):305-310.
14. Hodak E, Jones RE, Ackerman AB. Solitary keratoacanthoma is a squamous-cell carcinoma: three examples with metastases. Am J Dermatopathol. 1993;15(4):332-342.
15. Hofer SO, Jackson IT. Self-involution of giant keratoacanthoma on the tip of the nose. Plast Reconstr Surg. 2004;113(2):765-766.
16. Sung CS, Chuang FC, Ho JC, Lin SH. Keratoacanthoma centrifugum marginatum—A rare variant of keratoacanthoma: Case report and literature review. Dermatologica Sinica. 2014;32(1):25-28.
17. Schwartz RA. Keratoacanthoma. J Am Acad Dermatol. 1994;30(1):1-19; quiz 20-22.
18. Borkhatariya PB, Gupta S, Bang D, Rawal RC. Keratoacanthoma centrifugum marginatum: case report and review of literature. Indian J Dermatol. 2011;56(4):455-456.
19. Kwiek B, Schwartz RA. Keratoacanthoma (KA): An update and review. J Am Acad Dermatol. 2016;74(6):1220-1233.
20. Smith JF. Multiple primary, self-healing squamous epithelioma of the skin. Br J Dermatol. 1948;60(10):315-318.
21. Ponti G, Losi L, Di Gregorio C, et al. Identification of Muir-Torre syndrome among patients with sebaceous tumors and keratoacanthomas: role of clinical features, microsatellite instability, and immunohistochemistry. Cancer. 2005;103(5):1018-1025.
22. Agarwal M, Chander R, Karmakar S, Walia R. Multiple familial keratoacanthoma of Witten and Zak–a report of three siblings. Dermatology. 1999;198(4):396-399.
23. Ansai S, Manabe M. Possible spontaneous regression of a metastatic lesion of keratoacanthoma-like squamous cell carcinoma in a regional lymph node. J Dermatol. 2005;32(11):899-903.
24. Davis BA, Monheit GD, Kline L. Metastatic skin cancer presenting as ptosis and diplopia. Dermatol Surg. 2006;32(1):148-158.
25. Patrie M, Yehuda E, Carmen C. Keratoacanthoma of the head and neck with perineural invasion: incidental finding or cause for concern? Dermatol Surg. 2010;36(7):1209-1213.
26. Godbolt AM, Sullivan JJ, Weedon D. Keratoacanthoma with perineural invasion: a report of 40 cases. Australas J Dermatol. 2001;42(3):168-171.
27. Mandrell JC, Santa Cruz D. Keratoacanthoma: hyperplasia, benign neoplasm, or a type of squamous cell carcinoma? Semin Diagn Pathol. 2009;26(3):150-163.
28. Putti TC, Teh M, Lee YS. Biological behavior of keratoacanthoma and squamous cell carcinoma: telomerase activity and COX-2 as potential markers. Mod Pathol. 2004;17(4):468-475.
29. Weedon DD, Malo J, Brooks D, Williamson R. Squamous cell carcinoma arising in keratoacanthoma: a neglected phenomenon in the elderly. Am J Dermatopathol. 2010;32(5):423-426.
30. Vandergriff T, Nakamura K, High WA. Generalized eruptive keratoacanthomas of Grzybowski treated with isotretinoin. J Drugs Dermatol. 2008;7(11):1069-1071.



















