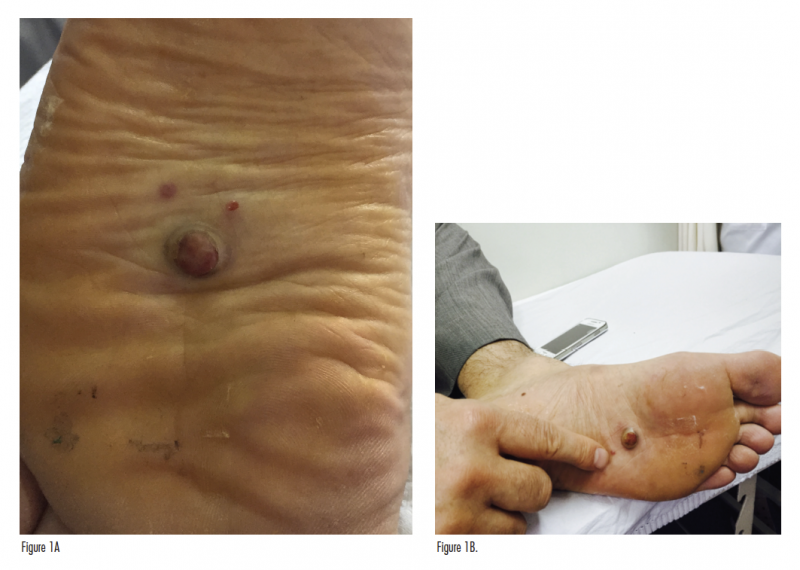
An otherwise healthy 43-year-old man presented with newly developed lesions on the plantar surface of the left foot. During a full-body skin examination, we noted a red papule and a well-circumscribed, nontender, firm, violaceous to dark-red nodule bordered by an epidermal collarette on the plantar surface of the left foot. The nodular and the papular lesions measured 1.5 × 1.5 cm and 0.3 × 0.3 cm, respectively (Figures 1A and 1B). The patient reported that these lesions have been present for 20 days, have been bleeding easily, and have been increasing in size, but denied other symptoms such as pain or pruritus.
What is Your Diagnosis?
Answer on page 2
{{pagebreak}}
Diagnosis: Kaposi Sarcoma Mimicking Pyogenic Granuloma
Kaposi sarcoma (KS) is a tumor characterized by neoplastic proliferation of endothelial cells.1 KS was described in 1872 by Moritz Kaposi as a new entity.2 Subsequently, other researchers described 4 clinical subtypes of KS that had identical histologic features but developed in particular people and had different involvement.3 These 4 recognized clinical variants include: classic, epidemic, iatrogenic, and endemic. Human herpesvirus 8 (HHV-8) has been established as the causative agent in all types of KS.4
Clinical Presentation of Kaposi Sarcoma
KS is a multifocal neoplasm that generally presents as multiple vascular cutaneous and mucosal lesions.5 Many factors vary with the subtype of KS, including the number of skin lesions, the favored location, the possibility of mucosal and visceral involvement, and the prognosis of the disease. Its course ranges from only cutaneous lesions often limited to the lower extremities to extensive cutaneous and visceral disease.1 In the classic type, lesions occur predominantly on the feet and distal part of legs, especially around the ankles.2 Lesions may start as millimetric purple, red, or flesh-colored macules or papules. These may enlarge and become large plaques or nodules.6 The lesions may be flattened, hyperkeratotic, verrucous, eroded or ulcerated, and covered with crusts.1 Large tumors may cause bleeding and pain. Infrequently, they may appear as subcutaneous nodules7 or develop telangiectasia on the surface.8
Sometimes it may present as ecchymoses on the periorbital skin.9 Keloidal nodules on KS lesions may appear in patients who have an underlying predisposition to keloid formation.10 KS mimicking lesions of chilblains on the hands11 or with pyogenic granuloma-like appearance have been reported.12 Depending on the extent of lesions, lymphedema can occur as a complication.13
Unlike the classic form, epidemic KS, seen in patients with HIV infection, has an aggressive course.14 In addition to the distal extremities, widely distributed skin lesions occur on the head, neck, upper trunk, and oral/genital mucosa.6 Lymph node and visceral involvement is more frequent than in the classic type.15 Furthermore, immunocompromised patients secondary to organ transplantation, systemic lymphomas, or long-term systemic immunosuppressive administration usually have multiple lesions.1 Lesions are primarily located on the distal extremities as in the classic form, but also on the head, trunk, proximal extremities, mucosa, and visceral organs.6
In African endemic-KS, the distal extremities are mainly involved and systemic involvement is not common, as in the classic subtype. On the other hand, the endemic subtype is locally more aggressive.15
Epidemiology
Classic KS is the most common form of the tumor in many countries, excluding those where the endemic form is generally seen.3 It is common in people of Mediterranean and Eastern European Jewish origin. The incidence of classic KS peaks after the sixth decade of life, with an obvious male predominance.16 Epidemic subtype is more commonly seen in HIV-positive men, with the risk of developing KS in these patients being 20,000 times higher than the general population.17 On the other hand, the incidence and morbidity are dramatically reduced after the use of highly activate antiretroviral therapy.18 In immunocompromised patients, lesions may appear at the beginning of immunosuppression or up to 10 years later, with men more often affected than women. Endemic KS is usually seen in African black men and children in some African countries near the equator.16
Article continues on page 3
{{pagebreak}}
Etiopathogenesis and Histopathology
KS is a tumor that is caused by an excessive proliferation of spindle cells thought to have a vascular or lymphatic endothelial cell origin.19 These spindle cells express pan-endothelial markers like CD31, blood vascular differentiation markers like CD34, and lymphatic differentiation markers like VEGFR-3, podoplanin, and LYVE-1.20,21 HHV-8 plays an important role in the etiopathogenesis as it may infect both lymphatic and blood vascular endothelial cells, stimulate a clonal proliferation of tumor cells, cause inflammation, and induce angiogenesis but inhibit apoptotic processes.22
The histologic appearance of KS is identical between the different clinical subtypes, but varies with stage of the lesion. The early plaque lesions may show mild changes such as slit-like abnormal vascular spaces, small numbers of spindle cells expressing endothelial markers, a reactive inflammatory infiltrate containing plasma cells, and hemosiderin deposition related with erythrocyte extravasation.23 In the advanced plaque lesions, the vascular proliferation extends to the deeper dermis and even the subcutis. In the nodular phase, dermal collagen is replaced by spindled endothelial cells.24 There is no pleomorphism or significant numbers of mitotic figures, and immunostaining for HHV-8 is always positive.25 Furthermore, the tumors stain positively for immunohistochemical markers of both lymphoid, spindle, and endothelial cells.20
Differential Diagnosis
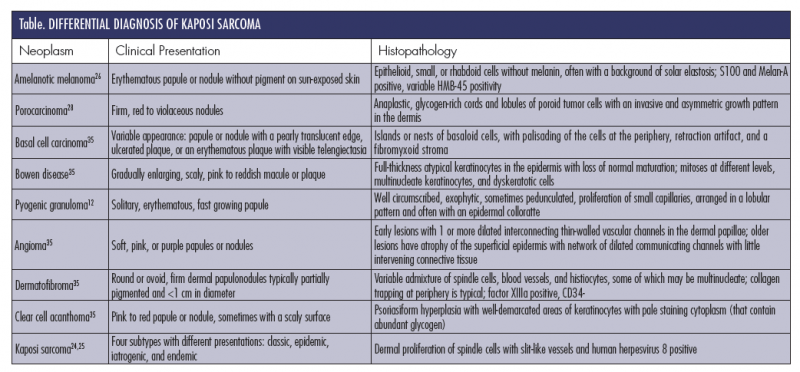
The differential diagnosis of KS (Table) is broad and includes a number of malignant and benign neoplasms. A plantar location and clinical features may help to narrow the differential diagnosis to pyogenic granuloma, amelanotic melanoma, KS, and eccrine poroma. Dermoscopy may support a physician’s clinical suspicion. However, histopathology is often necessary for diagnosis. As such, when KS is suspected clinically, biopsy is recommended to rule out other neoplasms. Immunostaining for HHV- 8 LNA-1 (latent nuclear antigen-1) helps distinguish KS from other differential diagnoses.25
Pyogenic granuloma appears clinically as a dome-shaped lesion bordered by an epidermal collarette, most commonly on the extremities. Histologically, pyogenic granulomas consist of a lobular proliferation of capillary vessels in the dermis. In contrast, KS has a dense proliferation of spindle cells arranged in fascicles that is often associated with an inflammatory cell infiltrate, which is not seen in pyogenic granuloma.4,12 Clinically, pyogenic granuloma is usually a solitary lesion, whereas KS most commonly presents as a group of lesions.
Amelanotic melanoma accounts for 2% to 8% of all melanomas. Because it usually presents as a vascular nodule, rather than as a pigmented nevus, the lesion may be misdiagnosed for a benign lesion. The clinical diagnostic features normally associated with melanomas, such as asymmetry, irregular borders, and color variation, are rarely seen in the amelanotic melanomas.26 For that reason, histopathology and immunohistochemistry are essential for diagnosis. Immunostaining with melanotic markers like S100 and HMB45 helps differentiate amelanotic melanoma from KS.27
The clinical features of eccrine poroma and lesion location may show similarity with KS. Poromas usually present as a dome-shaped, red, solitary nodule on the soles or sides of the foot.28 They may have a thickened collar of epidermis as in pyogenic granulomas.6 The lesions are diagnosed histologically, where the tumor appears within the epidermis and extends into the dermis. Lobular proliferation of monomorphic cuboidal small cells and scattered duct-like structures are seen on histopathologic examination.29
Treatment
Identifying the subtype of KS is the first step in selecting the ideal therapeutic approach. Several local therapeutic modalities exist. Solitary lesions may be excised surgically or destroyed with lasers or cryotherapy.30 Intralesional vinca alkaloids, interferon-alpha, and topical retinoids are other options for limited lesions.5 For larger skin lesions, radiotherapy may be an effective treatment.31 Lesions that are refractory to other therapies, rapidly progressive, and accompanied by complications or visceral involvement need to be treated with systemic chemotherapy such as vinblastine, liposomal doxorubicin, liposomal daunorubicin, paclitaxel, and etoposide.32 Systemic and local therapies may be combined in some cases. In AIDS-associated KS, the disseminated skin lesions may regress with antiretroviral therapy.33 Similarly, if possible, withdrawal of immunosuppressive agent is effective in the management of the iatrogenic subtype of KS and may result in complete resolution.34
Our Patient
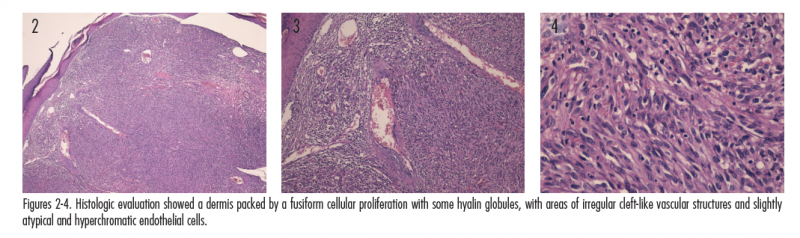
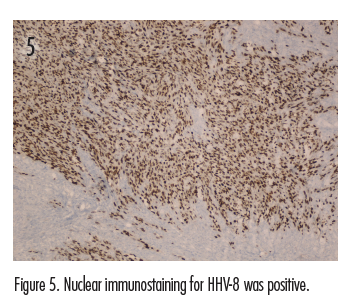
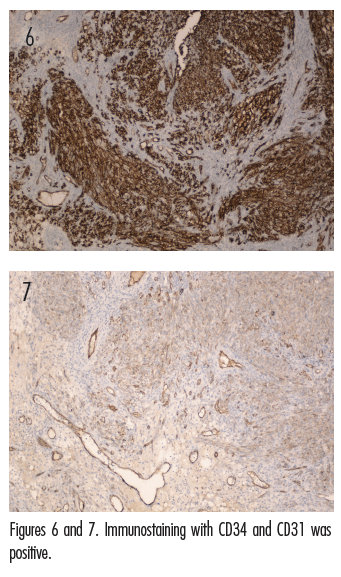
The initial clinical impression in our patient favored pyogenic granuloma given the morphologic appearance, acral location, and history of KS due to the existence of satellite lesion (Figures 1A and 1B); however, other neoplasms, including KS, amelanotic melanoma, or eccrine poroma could not be ruled out. Therefore, an excisional biopsy of the lesion was performed. Histologic evaluation showed a dermis packed by a fusiform cellular proliferation with some hyalin globules, with areas of irregular cleft-like vascular structures and slightly atypical and hyperchromatic endothelial cells (Figures 2-4). Nuclear immunostaining for HHV-8 was positive (Figure 5). Additionally, immunostaining with CD34 and CD31 was positive (Figures 6 and 7). These features were consistent with a diagnosis of KS. The lesion was surgically excised with good healing. The patient is being followed at our clinic. At his most recent visit, the patient did not show any signs of recurrence.
Conclusion
Our case represents a pyogenic granuloma-like manifestation of KS. Pyogenic granuloma-like KS is a new entity that exhibits the clinical features of pyogenic granuloma and the histopathologic features of KS.4 Biopsy is necessary in confirming the diagnosis. The histopathology and immunohistochemical studies of our case confirmed the diagnosis of KS.
 Dr Örnek is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Dr Örnek is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Dr Kocatürk is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Dr Erdem is with Okmeydanı Training and Research Hospital, Department of Pathology, in Istanbul, Turkey.
Dr Özdemir is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Dr Duman is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Disclosure: The authors report no relevant financial relationships.
References
1. Schwarts RA, Micali G, Nasca MR, Scuderi L. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol. 2008;59(2):179-206.
2. Laresche C, Fournier E, Dupond AS, et al. Kaposi’s sarcoma: a population-based cancer registry descriptive study of 57 consecutive cases diagnosed between 1977 and 2009. Int J Dermatol. 2014;53(12):e549-e554.
An otherwise healthy 43-year-old man presented with newly developed lesions on the plantar surface of the left foot. During a full-body skin examination, we noted a red papule and a well-circumscribed, nontender, firm, violaceous to dark-red nodule bordered by an epidermal collarette on the plantar surface of the left foot. The nodular and the papular lesions measured 1.5 × 1.5 cm and 0.3 × 0.3 cm, respectively (Figures 1A and 1B). The patient reported that these lesions have been present for 20 days, have been bleeding easily, and have been increasing in size, but denied other symptoms such as pain or pruritus.

What is Your Diagnosis?
Diagnosis: Kaposi Sarcoma Mimicking Pyogenic Granuloma
Kaposi sarcoma (KS) is a tumor characterized by neoplastic proliferation of endothelial cells.1 KS was described in 1872 by Moritz Kaposi as a new entity.2 Subsequently, other researchers described 4 clinical subtypes of KS that had identical histologic features but developed in particular people and had different involvement.3 These 4 recognized clinical variants include: classic, epidemic, iatrogenic, and endemic. Human herpesvirus 8 (HHV-8) has been established as the causative agent in all types of KS.4
Clinical Presentation of Kaposi Sarcoma
KS is a multifocal neoplasm that generally presents as multiple vascular cutaneous and mucosal lesions.5 Many factors vary with the subtype of KS, including the number of skin lesions, the favored location, the possibility of mucosal and visceral involvement, and the prognosis of the disease. Its course ranges from only cutaneous lesions often limited to the lower extremities to extensive cutaneous and visceral disease.1 In the classic type, lesions occur predominantly on the feet and distal part of legs, especially around the ankles.2 Lesions may start as millimetric purple, red, or flesh-colored macules or papules. These may enlarge and become large plaques or nodules.6 The lesions may be flattened, hyperkeratotic, verrucous, eroded or ulcerated, and covered with crusts.1 Large tumors may cause bleeding and pain. Infrequently, they may appear as subcutaneous nodules7 or develop telangiectasia on the surface.8
Sometimes it may present as ecchymoses on the periorbital skin.9 Keloidal nodules on KS lesions may appear in patients who have an underlying predisposition to keloid formation.10 KS mimicking lesions of chilblains on the hands11 or with pyogenic granuloma-like appearance have been reported.12 Depending on the extent of lesions, lymphedema can occur as a complication.13
Unlike the classic form, epidemic KS, seen in patients with HIV infection, has an aggressive course.14 In addition to the distal extremities, widely distributed skin lesions occur on the head, neck, upper trunk, and oral/genital mucosa.6 Lymph node and visceral involvement is more frequent than in the classic type.15 Furthermore, immunocompromised patients secondary to organ transplantation, systemic lymphomas, or long-term systemic immunosuppressive administration usually have multiple lesions.1 Lesions are primarily located on the distal extremities as in the classic form, but also on the head, trunk, proximal extremities, mucosa, and visceral organs.6
In African endemic-KS, the distal extremities are mainly involved and systemic involvement is not common, as in the classic subtype. On the other hand, the endemic subtype is locally more aggressive.15


Epidemiology
Classic KS is the most common form of the tumor in many countries, excluding those where the endemic form is generally seen.3 It is common in people of Mediterranean and Eastern European Jewish origin. The incidence of classic KS peaks after the sixth decade of life, with an obvious male predominance.16 Epidemic subtype is more commonly seen in HIV-positive men, with the risk of developing KS in these patients being 20,000 times higher than the general population.17 On the other hand, the incidence and morbidity are dramatically reduced after the use of highly activate antiretroviral therapy.18 In immunocompromised patients, lesions may appear at the beginning of immunosuppression or up to 10 years later, with men more often affected than women. Endemic KS is usually seen in African black men and children in some African countries near the equator.16
Article continues on page 3
{{pagebreak}}
Etiopathogenesis and Histopathology
KS is a tumor that is caused by an excessive proliferation of spindle cells thought to have a vascular or lymphatic endothelial cell origin.19 These spindle cells express pan-endothelial markers like CD31, blood vascular differentiation markers like CD34, and lymphatic differentiation markers like VEGFR-3, podoplanin, and LYVE-1.20,21 HHV-8 plays an important role in the etiopathogenesis as it may infect both lymphatic and blood vascular endothelial cells, stimulate a clonal proliferation of tumor cells, cause inflammation, and induce angiogenesis but inhibit apoptotic processes.22
The histologic appearance of KS is identical between the different clinical subtypes, but varies with stage of the lesion. The early plaque lesions may show mild changes such as slit-like abnormal vascular spaces, small numbers of spindle cells expressing endothelial markers, a reactive inflammatory infiltrate containing plasma cells, and hemosiderin deposition related with erythrocyte extravasation.23 In the advanced plaque lesions, the vascular proliferation extends to the deeper dermis and even the subcutis. In the nodular phase, dermal collagen is replaced by spindled endothelial cells.24 There is no pleomorphism or significant numbers of mitotic figures, and immunostaining for HHV-8 is always positive.25 Furthermore, the tumors stain positively for immunohistochemical markers of both lymphoid, spindle, and endothelial cells.20
Differential Diagnosis
The differential diagnosis of KS (Table) is broad and includes a number of malignant and benign neoplasms. A plantar location and clinical features may help to narrow the differential diagnosis to pyogenic granuloma, amelanotic melanoma, KS, and eccrine poroma. Dermoscopy may support a physician’s clinical suspicion. However, histopathology is often necessary for diagnosis. As such, when KS is suspected clinically, biopsy is recommended to rule out other neoplasms. Immunostaining for HHV- 8 LNA-1 (latent nuclear antigen-1) helps distinguish KS from other differential diagnoses.25
Pyogenic granuloma appears clinically as a dome-shaped lesion bordered by an epidermal collarette, most commonly on the extremities. Histologically, pyogenic granulomas consist of a lobular proliferation of capillary vessels in the dermis. In contrast, KS has a dense proliferation of spindle cells arranged in fascicles that is often associated with an inflammatory cell infiltrate, which is not seen in pyogenic granuloma.4,12 Clinically, pyogenic granuloma is usually a solitary lesion, whereas KS most commonly presents as a group of lesions.
Amelanotic melanoma accounts for 2% to 8% of all melanomas. Because it usually presents as a vascular nodule, rather than as a pigmented nevus, the lesion may be misdiagnosed for a benign lesion. The clinical diagnostic features normally associated with melanomas, such as asymmetry, irregular borders, and color variation, are rarely seen in the amelanotic melanomas.26 For that reason, histopathology and immunohistochemistry are essential for diagnosis. Immunostaining with melanotic markers like S100 and HMB45 helps differentiate amelanotic melanoma from KS.27
The clinical features of eccrine poroma and lesion location may show similarity with KS. Poromas usually present as a dome-shaped, red, solitary nodule on the soles or sides of the foot.28 They may have a thickened collar of epidermis as in pyogenic granulomas.6 The lesions are diagnosed histologically, where the tumor appears within the epidermis and extends into the dermis. Lobular proliferation of monomorphic cuboidal small cells and scattered duct-like structures are seen on histopathologic examination.29
Treatment
Identifying the subtype of KS is the first step in selecting the ideal therapeutic approach. Several local therapeutic modalities exist. Solitary lesions may be excised surgically or destroyed with lasers or cryotherapy.30 Intralesional vinca alkaloids, interferon-alpha, and topical retinoids are other options for limited lesions.5 For larger skin lesions, radiotherapy may be an effective treatment.31 Lesions that are refractory to other therapies, rapidly progressive, and accompanied by complications or visceral involvement need to be treated with systemic chemotherapy such as vinblastine, liposomal doxorubicin, liposomal daunorubicin, paclitaxel, and etoposide.32 Systemic and local therapies may be combined in some cases. In AIDS-associated KS, the disseminated skin lesions may regress with antiretroviral therapy.33 Similarly, if possible, withdrawal of immunosuppressive agent is effective in the management of the iatrogenic subtype of KS and may result in complete resolution.34


Our Patient
The initial clinical impression in our patient favored pyogenic granuloma given the morphologic appearance, acral location, and history of KS due to the existence of satellite lesion (Figures 1A and 1B); however, other neoplasms, including KS, amelanotic melanoma, or eccrine poroma could not be ruled out. Therefore, an excisional biopsy of the lesion was performed. Histologic evaluation showed a dermis packed by a fusiform cellular proliferation with some hyalin globules, with areas of irregular cleft-like vascular structures and slightly atypical and hyperchromatic endothelial cells (Figures 2-4). Nuclear immunostaining for HHV-8 was positive (Figure 5). Additionally, immunostaining with CD34 and CD31 was positive (Figures 6 and 7). These features were consistent with a diagnosis of KS. The lesion was surgically excised with good healing. The patient is being followed at our clinic. At his most recent visit, the patient did not show any signs of recurrence.
Conclusion
Our case represents a pyogenic granuloma-like manifestation of KS. Pyogenic granuloma-like KS is a new entity that exhibits the clinical features of pyogenic granuloma and the histopathologic features of KS.4 Biopsy is necessary in confirming the diagnosis. The histopathology and immunohistochemical studies of our case confirmed the diagnosis of KS.
 Dr Örnek is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Dr Örnek is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Dr Kocatürk is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Dr Erdem is with Okmeydanı Training and Research Hospital, Department of Pathology, in Istanbul, Turkey.
Dr Özdemir is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Dr Duman is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Disclosure: The authors report no relevant financial relationships.
References
1. Schwarts RA, Micali G, Nasca MR, Scuderi L. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol. 2008;59(2):179-206.
2. Laresche C, Fournier E, Dupond AS, et al. Kaposi’s sarcoma: a population-based cancer registry descriptive study of 57 consecutive cases diagnosed between 1977 and 2009. Int J Dermatol. 2014;53(12):e549-e554.
An otherwise healthy 43-year-old man presented with newly developed lesions on the plantar surface of the left foot. During a full-body skin examination, we noted a red papule and a well-circumscribed, nontender, firm, violaceous to dark-red nodule bordered by an epidermal collarette on the plantar surface of the left foot. The nodular and the papular lesions measured 1.5 × 1.5 cm and 0.3 × 0.3 cm, respectively (Figures 1A and 1B). The patient reported that these lesions have been present for 20 days, have been bleeding easily, and have been increasing in size, but denied other symptoms such as pain or pruritus.
What is Your Diagnosis?
,
An otherwise healthy 43-year-old man presented with newly developed lesions on the plantar surface of the left foot. During a full-body skin examination, we noted a red papule and a well-circumscribed, nontender, firm, violaceous to dark-red nodule bordered by an epidermal collarette on the plantar surface of the left foot. The nodular and the papular lesions measured 1.5 × 1.5 cm and 0.3 × 0.3 cm, respectively (Figures 1A and 1B). The patient reported that these lesions have been present for 20 days, have been bleeding easily, and have been increasing in size, but denied other symptoms such as pain or pruritus.
What is Your Diagnosis?
Answer on page 2
{{pagebreak}}
Diagnosis: Kaposi Sarcoma Mimicking Pyogenic Granuloma
Kaposi sarcoma (KS) is a tumor characterized by neoplastic proliferation of endothelial cells.1 KS was described in 1872 by Moritz Kaposi as a new entity.2 Subsequently, other researchers described 4 clinical subtypes of KS that had identical histologic features but developed in particular people and had different involvement.3 These 4 recognized clinical variants include: classic, epidemic, iatrogenic, and endemic. Human herpesvirus 8 (HHV-8) has been established as the causative agent in all types of KS.4
Clinical Presentation of Kaposi Sarcoma
KS is a multifocal neoplasm that generally presents as multiple vascular cutaneous and mucosal lesions.5 Many factors vary with the subtype of KS, including the number of skin lesions, the favored location, the possibility of mucosal and visceral involvement, and the prognosis of the disease. Its course ranges from only cutaneous lesions often limited to the lower extremities to extensive cutaneous and visceral disease.1 In the classic type, lesions occur predominantly on the feet and distal part of legs, especially around the ankles.2 Lesions may start as millimetric purple, red, or flesh-colored macules or papules. These may enlarge and become large plaques or nodules.6 The lesions may be flattened, hyperkeratotic, verrucous, eroded or ulcerated, and covered with crusts.1 Large tumors may cause bleeding and pain. Infrequently, they may appear as subcutaneous nodules7 or develop telangiectasia on the surface.8
Sometimes it may present as ecchymoses on the periorbital skin.9 Keloidal nodules on KS lesions may appear in patients who have an underlying predisposition to keloid formation.10 KS mimicking lesions of chilblains on the hands11 or with pyogenic granuloma-like appearance have been reported.12 Depending on the extent of lesions, lymphedema can occur as a complication.13
Unlike the classic form, epidemic KS, seen in patients with HIV infection, has an aggressive course.14 In addition to the distal extremities, widely distributed skin lesions occur on the head, neck, upper trunk, and oral/genital mucosa.6 Lymph node and visceral involvement is more frequent than in the classic type.15 Furthermore, immunocompromised patients secondary to organ transplantation, systemic lymphomas, or long-term systemic immunosuppressive administration usually have multiple lesions.1 Lesions are primarily located on the distal extremities as in the classic form, but also on the head, trunk, proximal extremities, mucosa, and visceral organs.6
In African endemic-KS, the distal extremities are mainly involved and systemic involvement is not common, as in the classic subtype. On the other hand, the endemic subtype is locally more aggressive.15
Epidemiology
Classic KS is the most common form of the tumor in many countries, excluding those where the endemic form is generally seen.3 It is common in people of Mediterranean and Eastern European Jewish origin. The incidence of classic KS peaks after the sixth decade of life, with an obvious male predominance.16 Epidemic subtype is more commonly seen in HIV-positive men, with the risk of developing KS in these patients being 20,000 times higher than the general population.17 On the other hand, the incidence and morbidity are dramatically reduced after the use of highly activate antiretroviral therapy.18 In immunocompromised patients, lesions may appear at the beginning of immunosuppression or up to 10 years later, with men more often affected than women. Endemic KS is usually seen in African black men and children in some African countries near the equator.16
Article continues on page 3
{{pagebreak}}
Etiopathogenesis and Histopathology
KS is a tumor that is caused by an excessive proliferation of spindle cells thought to have a vascular or lymphatic endothelial cell origin.19 These spindle cells express pan-endothelial markers like CD31, blood vascular differentiation markers like CD34, and lymphatic differentiation markers like VEGFR-3, podoplanin, and LYVE-1.20,21 HHV-8 plays an important role in the etiopathogenesis as it may infect both lymphatic and blood vascular endothelial cells, stimulate a clonal proliferation of tumor cells, cause inflammation, and induce angiogenesis but inhibit apoptotic processes.22
The histologic appearance of KS is identical between the different clinical subtypes, but varies with stage of the lesion. The early plaque lesions may show mild changes such as slit-like abnormal vascular spaces, small numbers of spindle cells expressing endothelial markers, a reactive inflammatory infiltrate containing plasma cells, and hemosiderin deposition related with erythrocyte extravasation.23 In the advanced plaque lesions, the vascular proliferation extends to the deeper dermis and even the subcutis. In the nodular phase, dermal collagen is replaced by spindled endothelial cells.24 There is no pleomorphism or significant numbers of mitotic figures, and immunostaining for HHV-8 is always positive.25 Furthermore, the tumors stain positively for immunohistochemical markers of both lymphoid, spindle, and endothelial cells.20
Differential Diagnosis
The differential diagnosis of KS (Table) is broad and includes a number of malignant and benign neoplasms. A plantar location and clinical features may help to narrow the differential diagnosis to pyogenic granuloma, amelanotic melanoma, KS, and eccrine poroma. Dermoscopy may support a physician’s clinical suspicion. However, histopathology is often necessary for diagnosis. As such, when KS is suspected clinically, biopsy is recommended to rule out other neoplasms. Immunostaining for HHV- 8 LNA-1 (latent nuclear antigen-1) helps distinguish KS from other differential diagnoses.25
Pyogenic granuloma appears clinically as a dome-shaped lesion bordered by an epidermal collarette, most commonly on the extremities. Histologically, pyogenic granulomas consist of a lobular proliferation of capillary vessels in the dermis. In contrast, KS has a dense proliferation of spindle cells arranged in fascicles that is often associated with an inflammatory cell infiltrate, which is not seen in pyogenic granuloma.4,12 Clinically, pyogenic granuloma is usually a solitary lesion, whereas KS most commonly presents as a group of lesions.
Amelanotic melanoma accounts for 2% to 8% of all melanomas. Because it usually presents as a vascular nodule, rather than as a pigmented nevus, the lesion may be misdiagnosed for a benign lesion. The clinical diagnostic features normally associated with melanomas, such as asymmetry, irregular borders, and color variation, are rarely seen in the amelanotic melanomas.26 For that reason, histopathology and immunohistochemistry are essential for diagnosis. Immunostaining with melanotic markers like S100 and HMB45 helps differentiate amelanotic melanoma from KS.27
The clinical features of eccrine poroma and lesion location may show similarity with KS. Poromas usually present as a dome-shaped, red, solitary nodule on the soles or sides of the foot.28 They may have a thickened collar of epidermis as in pyogenic granulomas.6 The lesions are diagnosed histologically, where the tumor appears within the epidermis and extends into the dermis. Lobular proliferation of monomorphic cuboidal small cells and scattered duct-like structures are seen on histopathologic examination.29
Treatment
Identifying the subtype of KS is the first step in selecting the ideal therapeutic approach. Several local therapeutic modalities exist. Solitary lesions may be excised surgically or destroyed with lasers or cryotherapy.30 Intralesional vinca alkaloids, interferon-alpha, and topical retinoids are other options for limited lesions.5 For larger skin lesions, radiotherapy may be an effective treatment.31 Lesions that are refractory to other therapies, rapidly progressive, and accompanied by complications or visceral involvement need to be treated with systemic chemotherapy such as vinblastine, liposomal doxorubicin, liposomal daunorubicin, paclitaxel, and etoposide.32 Systemic and local therapies may be combined in some cases. In AIDS-associated KS, the disseminated skin lesions may regress with antiretroviral therapy.33 Similarly, if possible, withdrawal of immunosuppressive agent is effective in the management of the iatrogenic subtype of KS and may result in complete resolution.34
Our Patient
The initial clinical impression in our patient favored pyogenic granuloma given the morphologic appearance, acral location, and history of KS due to the existence of satellite lesion (Figures 1A and 1B); however, other neoplasms, including KS, amelanotic melanoma, or eccrine poroma could not be ruled out. Therefore, an excisional biopsy of the lesion was performed. Histologic evaluation showed a dermis packed by a fusiform cellular proliferation with some hyalin globules, with areas of irregular cleft-like vascular structures and slightly atypical and hyperchromatic endothelial cells (Figures 2-4). Nuclear immunostaining for HHV-8 was positive (Figure 5). Additionally, immunostaining with CD34 and CD31 was positive (Figures 6 and 7). These features were consistent with a diagnosis of KS. The lesion was surgically excised with good healing. The patient is being followed at our clinic. At his most recent visit, the patient did not show any signs of recurrence.
Conclusion
Our case represents a pyogenic granuloma-like manifestation of KS. Pyogenic granuloma-like KS is a new entity that exhibits the clinical features of pyogenic granuloma and the histopathologic features of KS.4 Biopsy is necessary in confirming the diagnosis. The histopathology and immunohistochemical studies of our case confirmed the diagnosis of KS.
Dr Örnek is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Dr Kocatürk is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Dr Erdem is with Okmeydanı Training and Research Hospital, Department of Pathology, in Istanbul, Turkey.
Dr Özdemir is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Dr Duman is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Disclosure: The authors report no relevant financial relationships.
References
1. Schwarts RA, Micali G, Nasca MR, Scuderi L. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol. 2008;59(2):179-206.
2. Laresche C, Fournier E, Dupond AS, et al. Kaposi’s sarcoma: a population-based cancer registry descriptive study of 57 consecutive cases diagnosed between 1977 and 2009. Int J Dermatol. 2014;53(12):e549-e554.
An otherwise healthy 43-year-old man presented with newly developed lesions on the plantar surface of the left foot. During a full-body skin examination, we noted a red papule and a well-circumscribed, nontender, firm, violaceous to dark-red nodule bordered by an epidermal collarette on the plantar surface of the left foot. The nodular and the papular lesions measured 1.5 × 1.5 cm and 0.3 × 0.3 cm, respectively (Figures 1A and 1B). The patient reported that these lesions have been present for 20 days, have been bleeding easily, and have been increasing in size, but denied other symptoms such as pain or pruritus.
What is Your Diagnosis?
Diagnosis: Kaposi Sarcoma Mimicking Pyogenic Granuloma
Kaposi sarcoma (KS) is a tumor characterized by neoplastic proliferation of endothelial cells.1 KS was described in 1872 by Moritz Kaposi as a new entity.2 Subsequently, other researchers described 4 clinical subtypes of KS that had identical histologic features but developed in particular people and had different involvement.3 These 4 recognized clinical variants include: classic, epidemic, iatrogenic, and endemic. Human herpesvirus 8 (HHV-8) has been established as the causative agent in all types of KS.4
Clinical Presentation of Kaposi Sarcoma
KS is a multifocal neoplasm that generally presents as multiple vascular cutaneous and mucosal lesions.5 Many factors vary with the subtype of KS, including the number of skin lesions, the favored location, the possibility of mucosal and visceral involvement, and the prognosis of the disease. Its course ranges from only cutaneous lesions often limited to the lower extremities to extensive cutaneous and visceral disease.1 In the classic type, lesions occur predominantly on the feet and distal part of legs, especially around the ankles.2 Lesions may start as millimetric purple, red, or flesh-colored macules or papules. These may enlarge and become large plaques or nodules.6 The lesions may be flattened, hyperkeratotic, verrucous, eroded or ulcerated, and covered with crusts.1 Large tumors may cause bleeding and pain. Infrequently, they may appear as subcutaneous nodules7 or develop telangiectasia on the surface.8
Sometimes it may present as ecchymoses on the periorbital skin.9 Keloidal nodules on KS lesions may appear in patients who have an underlying predisposition to keloid formation.10 KS mimicking lesions of chilblains on the hands11 or with pyogenic granuloma-like appearance have been reported.12 Depending on the extent of lesions, lymphedema can occur as a complication.13
Unlike the classic form, epidemic KS, seen in patients with HIV infection, has an aggressive course.14 In addition to the distal extremities, widely distributed skin lesions occur on the head, neck, upper trunk, and oral/genital mucosa.6 Lymph node and visceral involvement is more frequent than in the classic type.15 Furthermore, immunocompromised patients secondary to organ transplantation, systemic lymphomas, or long-term systemic immunosuppressive administration usually have multiple lesions.1 Lesions are primarily located on the distal extremities as in the classic form, but also on the head, trunk, proximal extremities, mucosa, and visceral organs.6
In African endemic-KS, the distal extremities are mainly involved and systemic involvement is not common, as in the classic subtype. On the other hand, the endemic subtype is locally more aggressive.15
Epidemiology
Classic KS is the most common form of the tumor in many countries, excluding those where the endemic form is generally seen.3 It is common in people of Mediterranean and Eastern European Jewish origin. The incidence of classic KS peaks after the sixth decade of life, with an obvious male predominance.16 Epidemic subtype is more commonly seen in HIV-positive men, with the risk of developing KS in these patients being 20,000 times higher than the general population.17 On the other hand, the incidence and morbidity are dramatically reduced after the use of highly activate antiretroviral therapy.18 In immunocompromised patients, lesions may appear at the beginning of immunosuppression or up to 10 years later, with men more often affected than women. Endemic KS is usually seen in African black men and children in some African countries near the equator.16
Article continues on page 3
{{pagebreak}}
Etiopathogenesis and Histopathology
KS is a tumor that is caused by an excessive proliferation of spindle cells thought to have a vascular or lymphatic endothelial cell origin.19 These spindle cells express pan-endothelial markers like CD31, blood vascular differentiation markers like CD34, and lymphatic differentiation markers like VEGFR-3, podoplanin, and LYVE-1.20,21 HHV-8 plays an important role in the etiopathogenesis as it may infect both lymphatic and blood vascular endothelial cells, stimulate a clonal proliferation of tumor cells, cause inflammation, and induce angiogenesis but inhibit apoptotic processes.22
The histologic appearance of KS is identical between the different clinical subtypes, but varies with stage of the lesion. The early plaque lesions may show mild changes such as slit-like abnormal vascular spaces, small numbers of spindle cells expressing endothelial markers, a reactive inflammatory infiltrate containing plasma cells, and hemosiderin deposition related with erythrocyte extravasation.23 In the advanced plaque lesions, the vascular proliferation extends to the deeper dermis and even the subcutis. In the nodular phase, dermal collagen is replaced by spindled endothelial cells.24 There is no pleomorphism or significant numbers of mitotic figures, and immunostaining for HHV-8 is always positive.25 Furthermore, the tumors stain positively for immunohistochemical markers of both lymphoid, spindle, and endothelial cells.20
Differential Diagnosis
The differential diagnosis of KS (Table) is broad and includes a number of malignant and benign neoplasms. A plantar location and clinical features may help to narrow the differential diagnosis to pyogenic granuloma, amelanotic melanoma, KS, and eccrine poroma. Dermoscopy may support a physician’s clinical suspicion. However, histopathology is often necessary for diagnosis. As such, when KS is suspected clinically, biopsy is recommended to rule out other neoplasms. Immunostaining for HHV- 8 LNA-1 (latent nuclear antigen-1) helps distinguish KS from other differential diagnoses.25
Pyogenic granuloma appears clinically as a dome-shaped lesion bordered by an epidermal collarette, most commonly on the extremities. Histologically, pyogenic granulomas consist of a lobular proliferation of capillary vessels in the dermis. In contrast, KS has a dense proliferation of spindle cells arranged in fascicles that is often associated with an inflammatory cell infiltrate, which is not seen in pyogenic granuloma.4,12 Clinically, pyogenic granuloma is usually a solitary lesion, whereas KS most commonly presents as a group of lesions.
Amelanotic melanoma accounts for 2% to 8% of all melanomas. Because it usually presents as a vascular nodule, rather than as a pigmented nevus, the lesion may be misdiagnosed for a benign lesion. The clinical diagnostic features normally associated with melanomas, such as asymmetry, irregular borders, and color variation, are rarely seen in the amelanotic melanomas.26 For that reason, histopathology and immunohistochemistry are essential for diagnosis. Immunostaining with melanotic markers like S100 and HMB45 helps differentiate amelanotic melanoma from KS.27
The clinical features of eccrine poroma and lesion location may show similarity with KS. Poromas usually present as a dome-shaped, red, solitary nodule on the soles or sides of the foot.28 They may have a thickened collar of epidermis as in pyogenic granulomas.6 The lesions are diagnosed histologically, where the tumor appears within the epidermis and extends into the dermis. Lobular proliferation of monomorphic cuboidal small cells and scattered duct-like structures are seen on histopathologic examination.29
Treatment
Identifying the subtype of KS is the first step in selecting the ideal therapeutic approach. Several local therapeutic modalities exist. Solitary lesions may be excised surgically or destroyed with lasers or cryotherapy.30 Intralesional vinca alkaloids, interferon-alpha, and topical retinoids are other options for limited lesions.5 For larger skin lesions, radiotherapy may be an effective treatment.31 Lesions that are refractory to other therapies, rapidly progressive, and accompanied by complications or visceral involvement need to be treated with systemic chemotherapy such as vinblastine, liposomal doxorubicin, liposomal daunorubicin, paclitaxel, and etoposide.32 Systemic and local therapies may be combined in some cases. In AIDS-associated KS, the disseminated skin lesions may regress with antiretroviral therapy.33 Similarly, if possible, withdrawal of immunosuppressive agent is effective in the management of the iatrogenic subtype of KS and may result in complete resolution.34
Our Patient
The initial clinical impression in our patient favored pyogenic granuloma given the morphologic appearance, acral location, and history of KS due to the existence of satellite lesion (Figures 1A and 1B); however, other neoplasms, including KS, amelanotic melanoma, or eccrine poroma could not be ruled out. Therefore, an excisional biopsy of the lesion was performed. Histologic evaluation showed a dermis packed by a fusiform cellular proliferation with some hyalin globules, with areas of irregular cleft-like vascular structures and slightly atypical and hyperchromatic endothelial cells (Figures 2-4). Nuclear immunostaining for HHV-8 was positive (Figure 5). Additionally, immunostaining with CD34 and CD31 was positive (Figures 6 and 7). These features were consistent with a diagnosis of KS. The lesion was surgically excised with good healing. The patient is being followed at our clinic. At his most recent visit, the patient did not show any signs of recurrence.
Conclusion
Our case represents a pyogenic granuloma-like manifestation of KS. Pyogenic granuloma-like KS is a new entity that exhibits the clinical features of pyogenic granuloma and the histopathologic features of KS.4 Biopsy is necessary in confirming the diagnosis. The histopathology and immunohistochemical studies of our case confirmed the diagnosis of KS.
Dr Örnek is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Dr Kocatürk is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Dr Erdem is with Okmeydanı Training and Research Hospital, Department of Pathology, in Istanbul, Turkey.
Dr Özdemir is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Dr Duman is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Disclosure: The authors report no relevant financial relationships.
References
1. Schwarts RA, Micali G, Nasca MR, Scuderi L. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol. 2008;59(2):179-206.
2. Laresche C, Fournier E, Dupond AS, et al. Kaposi’s sarcoma: a population-based cancer registry descriptive study of 57 consecutive cases diagnosed between 1977 and 2009. Int J Dermatol. 2014;53(12):e549-e554.
Diagnosis: Kaposi Sarcoma Mimicking Pyogenic Granuloma
Kaposi sarcoma (KS) is a tumor characterized by neoplastic proliferation of endothelial cells.1 KS was described in 1872 by Moritz Kaposi as a new entity.2 Subsequently, other researchers described 4 clinical subtypes of KS that had identical histologic features but developed in particular people and had different involvement.3 These 4 recognized clinical variants include: classic, epidemic, iatrogenic, and endemic. Human herpesvirus 8 (HHV-8) has been established as the causative agent in all types of KS.4
Clinical Presentation of Kaposi Sarcoma
KS is a multifocal neoplasm that generally presents as multiple vascular cutaneous and mucosal lesions.5 Many factors vary with the subtype of KS, including the number of skin lesions, the favored location, the possibility of mucosal and visceral involvement, and the prognosis of the disease. Its course ranges from only cutaneous lesions often limited to the lower extremities to extensive cutaneous and visceral disease.1 In the classic type, lesions occur predominantly on the feet and distal part of legs, especially around the ankles.2 Lesions may start as millimetric purple, red, or flesh-colored macules or papules. These may enlarge and become large plaques or nodules.6 The lesions may be flattened, hyperkeratotic, verrucous, eroded or ulcerated, and covered with crusts.1 Large tumors may cause bleeding and pain. Infrequently, they may appear as subcutaneous nodules7 or develop telangiectasia on the surface.8
Sometimes it may present as ecchymoses on the periorbital skin.9 Keloidal nodules on KS lesions may appear in patients who have an underlying predisposition to keloid formation.10 KS mimicking lesions of chilblains on the hands11 or with pyogenic granuloma-like appearance have been reported.12 Depending on the extent of lesions, lymphedema can occur as a complication.13
Unlike the classic form, epidemic KS, seen in patients with HIV infection, has an aggressive course.14 In addition to the distal extremities, widely distributed skin lesions occur on the head, neck, upper trunk, and oral/genital mucosa.6 Lymph node and visceral involvement is more frequent than in the classic type.15 Furthermore, immunocompromised patients secondary to organ transplantation, systemic lymphomas, or long-term systemic immunosuppressive administration usually have multiple lesions.1 Lesions are primarily located on the distal extremities as in the classic form, but also on the head, trunk, proximal extremities, mucosa, and visceral organs.6
In African endemic-KS, the distal extremities are mainly involved and systemic involvement is not common, as in the classic subtype. On the other hand, the endemic subtype is locally more aggressive.15
Epidemiology
Classic KS is the most common form of the tumor in many countries, excluding those where the endemic form is generally seen.3 It is common in people of Mediterranean and Eastern European Jewish origin. The incidence of classic KS peaks after the sixth decade of life, with an obvious male predominance.16 Epidemic subtype is more commonly seen in HIV-positive men, with the risk of developing KS in these patients being 20,000 times higher than the general population.17 On the other hand, the incidence and morbidity are dramatically reduced after the use of highly activate antiretroviral therapy.18 In immunocompromised patients, lesions may appear at the beginning of immunosuppression or up to 10 years later, with men more often affected than women. Endemic KS is usually seen in African black men and children in some African countries near the equator.16
Article continues on page 3
{{pagebreak}}
Etiopathogenesis and Histopathology
KS is a tumor that is caused by an excessive proliferation of spindle cells thought to have a vascular or lymphatic endothelial cell origin.19 These spindle cells express pan-endothelial markers like CD31, blood vascular differentiation markers like CD34, and lymphatic differentiation markers like VEGFR-3, podoplanin, and LYVE-1.20,21 HHV-8 plays an important role in the etiopathogenesis as it may infect both lymphatic and blood vascular endothelial cells, stimulate a clonal proliferation of tumor cells, cause inflammation, and induce angiogenesis but inhibit apoptotic processes.22
The histologic appearance of KS is identical between the different clinical subtypes, but varies with stage of the lesion. The early plaque lesions may show mild changes such as slit-like abnormal vascular spaces, small numbers of spindle cells expressing endothelial markers, a reactive inflammatory infiltrate containing plasma cells, and hemosiderin deposition related with erythrocyte extravasation.23 In the advanced plaque lesions, the vascular proliferation extends to the deeper dermis and even the subcutis. In the nodular phase, dermal collagen is replaced by spindled endothelial cells.24 There is no pleomorphism or significant numbers of mitotic figures, and immunostaining for HHV-8 is always positive.25 Furthermore, the tumors stain positively for immunohistochemical markers of both lymphoid, spindle, and endothelial cells.20
Differential Diagnosis
The differential diagnosis of KS (Table) is broad and includes a number of malignant and benign neoplasms. A plantar location and clinical features may help to narrow the differential diagnosis to pyogenic granuloma, amelanotic melanoma, KS, and eccrine poroma. Dermoscopy may support a physician’s clinical suspicion. However, histopathology is often necessary for diagnosis. As such, when KS is suspected clinically, biopsy is recommended to rule out other neoplasms. Immunostaining for HHV- 8 LNA-1 (latent nuclear antigen-1) helps distinguish KS from other differential diagnoses.25
Pyogenic granuloma appears clinically as a dome-shaped lesion bordered by an epidermal collarette, most commonly on the extremities. Histologically, pyogenic granulomas consist of a lobular proliferation of capillary vessels in the dermis. In contrast, KS has a dense proliferation of spindle cells arranged in fascicles that is often associated with an inflammatory cell infiltrate, which is not seen in pyogenic granuloma.4,12 Clinically, pyogenic granuloma is usually a solitary lesion, whereas KS most commonly presents as a group of lesions.
Amelanotic melanoma accounts for 2% to 8% of all melanomas. Because it usually presents as a vascular nodule, rather than as a pigmented nevus, the lesion may be misdiagnosed for a benign lesion. The clinical diagnostic features normally associated with melanomas, such as asymmetry, irregular borders, and color variation, are rarely seen in the amelanotic melanomas.26 For that reason, histopathology and immunohistochemistry are essential for diagnosis. Immunostaining with melanotic markers like S100 and HMB45 helps differentiate amelanotic melanoma from KS.27
The clinical features of eccrine poroma and lesion location may show similarity with KS. Poromas usually present as a dome-shaped, red, solitary nodule on the soles or sides of the foot.28 They may have a thickened collar of epidermis as in pyogenic granulomas.6 The lesions are diagnosed histologically, where the tumor appears within the epidermis and extends into the dermis. Lobular proliferation of monomorphic cuboidal small cells and scattered duct-like structures are seen on histopathologic examination.29
Treatment
Identifying the subtype of KS is the first step in selecting the ideal therapeutic approach. Several local therapeutic modalities exist. Solitary lesions may be excised surgically or destroyed with lasers or cryotherapy.30 Intralesional vinca alkaloids, interferon-alpha, and topical retinoids are other options for limited lesions.5 For larger skin lesions, radiotherapy may be an effective treatment.31 Lesions that are refractory to other therapies, rapidly progressive, and accompanied by complications or visceral involvement need to be treated with systemic chemotherapy such as vinblastine, liposomal doxorubicin, liposomal daunorubicin, paclitaxel, and etoposide.32 Systemic and local therapies may be combined in some cases. In AIDS-associated KS, the disseminated skin lesions may regress with antiretroviral therapy.33 Similarly, if possible, withdrawal of immunosuppressive agent is effective in the management of the iatrogenic subtype of KS and may result in complete resolution.34
Our Patient
The initial clinical impression in our patient favored pyogenic granuloma given the morphologic appearance, acral location, and history of KS due to the existence of satellite lesion (Figures 1A and 1B); however, other neoplasms, including KS, amelanotic melanoma, or eccrine poroma could not be ruled out. Therefore, an excisional biopsy of the lesion was performed. Histologic evaluation showed a dermis packed by a fusiform cellular proliferation with some hyalin globules, with areas of irregular cleft-like vascular structures and slightly atypical and hyperchromatic endothelial cells (Figures 2-4). Nuclear immunostaining for HHV-8 was positive (Figure 5). Additionally, immunostaining with CD34 and CD31 was positive (Figures 6 and 7). These features were consistent with a diagnosis of KS. The lesion was surgically excised with good healing. The patient is being followed at our clinic. At his most recent visit, the patient did not show any signs of recurrence.
Conclusion
Our case represents a pyogenic granuloma-like manifestation of KS. Pyogenic granuloma-like KS is a new entity that exhibits the clinical features of pyogenic granuloma and the histopathologic features of KS.4 Biopsy is necessary in confirming the diagnosis. The histopathology and immunohistochemical studies of our case confirmed the diagnosis of KS.
Dr Örnek is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Dr Kocatürk is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Dr Erdem is with Okmeydanı Training and Research Hospital, Department of Pathology, in Istanbul, Turkey.
Dr Özdemir is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Dr Duman is with Okmeydanı Training and Research Hospital, Department of Dermatology, in Istanbul, Turkey.
Disclosure: The authors report no relevant financial relationships.
References
1. Schwarts RA, Micali G, Nasca MR, Scuderi L. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol. 2008;59(2):179-206.
2. Laresche C, Fournier E, Dupond AS, et al. Kaposi’s sarcoma: a population-based cancer registry descriptive study of 57 consecutive cases diagnosed between 1977 and 2009. Int J Dermatol. 2014;53(12):e549-e554.
















