This case is reminiscent of Sézary syndrome as the defining histomorphology in a patient with stage 1B mycosis fungoides: A clue to early peripheral blood involvement.
A 36-year-old woman presented to her primary care physician with pruritic rash localized to her inner thighs of 7 months’ duration. She had a history of multiple miscarriages, and therefore had peripheral blood cytogenetics and extensive autoimmune work-up from 2014 that were negative. Despite applying triamcinolone topical cream and taking hydroxyzine for 1 month, her pruritus worsened and extended to the hands and feet. Overall, the extent of pruritus was seemingly disparate to the degree of cutaneous disease. Physical examination revealed additional sites of involvement including arms, abdomen, and back. Hyperkeratosis was observed on the palms and soles.

Histopathologic Features
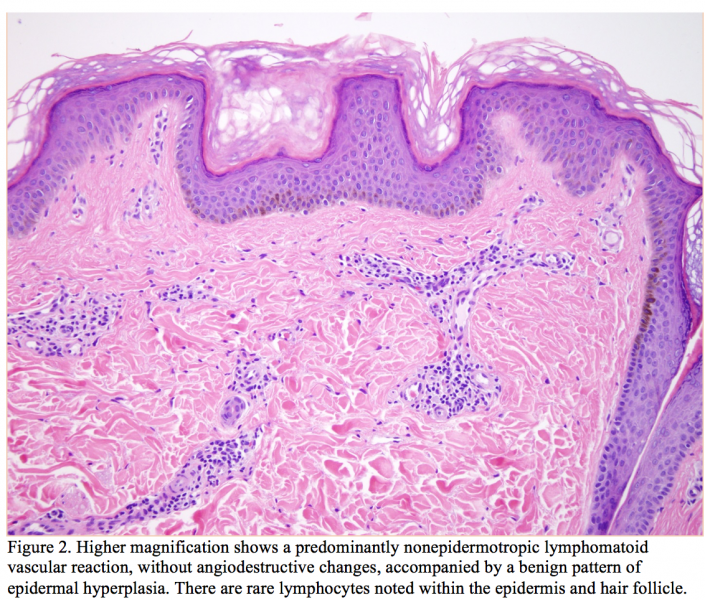
Punch biopsies taken from the bilateral arms revealed a predominantly angiocentric atypical lymphocytic infiltrate in the superficial and mid-dermis with minimal epitheliotropism (Figures 1 and 2). There was some degree of permeation of the interstitium superficially with attendant subepidermal fibrosis. The infiltrate was predominated by small- to intermediate-sized lymphocytes showing nuclear contour irregularity including many cells with hyperconvoluted cerebriform nuclei (Figure 3). A few plasma cells were identified as well. There was a concomitant mild epidermal hyperplasia surmounted by an orthohyperkeratotic scale. While the infiltrate was not significantly epidermotropic, there was very focal folliculotropism, whereby intrafollicular collections of cerebriform lymphocyte defining an interfollicular Pautrier microabscess were identified.



Immunohistochemical Findings
Comprehensive phenotypic studies were performed. The lymphocytic infiltrate was extensively highlighted by CD3 and pan T-cell marker CD2. The CD4 to CD8 ratio showed a clear-cut predominance of CD4 T cells over those of the CD8 subset with a CD4 to CD8 ratio exceeding 5:1 (Figures 4 and 5). The lymphocytes were of the alpha beta subset based on the extent of immunoreactivity for ß-F1. Striking and extensive immunoreactivity of the lymphocytes for programmed death 1 (PD-1) was noted (Figure 6). Marked upregulation of TOX amidst the lymphocytes (Figure 7) including strong nuclear expression of TOX amidst angiocentric lymphocytes was observed. There was focal staining for CD25 (20%-30% of the infiltrate) and the CD7 preparation showed a significant decrement in staining with only 20% of the infiltrate exhibiting positivity (Figure 8). There were a few FOXP3 positive staining T cells, but the majority of the cells did not exhibit a regulatory T-cell phenotype. Overall, the combined light microscopic findings and phenotypic profile was highly suggestive of Sézary syndrome.



Peripheral Blood Studies
She subsequently had peripheral blood studies performed. Her studies disclosed a small percentage of abnormal T cells averaging 0.25% of the entire population examined exhibiting expression of CD2, CD3, and CD4 but with loss of CD5 and CD7. The peripheral blood studies were interpreted as being reflective of minimal involvement by the patient’s cutaneous lymphoma.
Discussion
Sézary syndrome is characterized by the triad of erythroderma, lymphadenopathy, and abnormal T cells (Sézary cells) in the peripheral blood, lymph nodes, and skin. The authors of the 2005 World Health Organization-European Organization for Research and Treatment of Cancer classification for this cutaneous lymphoma suggested that there must be an absolute criterion, which is one of established T-cell clonality in the peripheral blood and skin, ideally demonstrating the same T-cell clone in the 2 different anatomic samples.1 The authors proposed 2 additional criteria, of which at least one must be present. The first is both cytomorphologic and quantitative, requiring a count of 1000 or more Sézary cells per square millimeter in the peripheral blood. The second criterion is immunophenotypic; the CD4 to CD8 T-lymphocyte ratio in the peripheral blood must be in excess of 10:1, with a concomitant loss of pan T-cell markers such as CD2, CD3, CD7, and CD5.1,2
The histopathology was not characteristic for mycosis fungoides as it was a predominantly angiocentric atypical lymphocytic infiltrate with minimal epitheliotropism. This particular morphology and phenotype characterized by a superficial perivascular atypical cerebriform CD4+ PD-1+, and CD7-lymphocytic infiltrate is quite characteristic for Sézary syndrome. Unlike mycosis fungoides where one expects to see conspicuous epidermotropism, including Pautrier microabscesses, biopsies in the setting of Sézary syndrome may be minimally epidermotropic. The infiltrate of Sézary syndrome lies largely within the dermis, exhibiting a vasocentric disposition around vessels of the superficial vascular plexus.
However, this patient did not fulfill criteria for Sézary syndrome based on peripheral blood assessment and lack of erythroderma. This patient was seen by a cutaneous lymphoma expert and was diagnosed as having mycosis fungoides stage 1B. One could speculate that the Sézary syndrome-like biopsy findings predicted peripheral blood involvement albeit not of the magnitude to be considered diagnostic of Sézary syndrome. In this regard, the distinctive histology suggesting a diagnosis of Sézary syndrome could also signify a subset of mycosis fungoides patients who have the potential for a more aggressive clinical course specifically as it applies to peripheral blood involvement.
The expression in this case of PD-1 is a critical phenotypic determinant in the setting of Sézary syndrome. PD-1 is an inhibitory member of the CD28/CTLA-4 family. The PD-1 pathway exerts its function through inhibiting TCR-mediated T-cell proliferation and cytokine production. PD-1 has been useful in differentiating Sézary syndrome from erythrodermic inflammatory dermatoses when expressed in CD4+ T cells over CD8+ T cells.3 Biopsies of Sézary syndrome typically show very prominent expression of PD-1 that far exceeds that in mycosis fungoides.4
It has postulated that Sézary syndrome is a neoplasm of central memory T cells and differs from conventional mycosis fungoides in which the cell of origin is held to be a resident effector T cell. Due to the abnormal pattern of PD-1 staining, a reactive based etiology including in the context of a lymphomatoid drug reaction is not favored.
In summary, the biopsies show a PD-1 and CD4 positive lymphomatoid vascular reaction most suggestive of Sézary syndrome but with clinical features of stage 1B mycosis fungoides and not Sézary syndrome. It is possible that this limited cutaneous presentation along minimal blood involvement may correspond with the earliest manifestation of Sézary syndrome. Conversely this distinct histomorphologic and phenotypic pattern could predict mycosis fungoides patients who may develop peripheral blood involvement with the potential for disease progression to Sézary syndrome.
 Dr Magro is the director of dermatopathology at Weill Cornell Medicine in New York, NY.
Dr Magro is the director of dermatopathology at Weill Cornell Medicine in New York, NY.
Dr Tsang is a fellow in transfusion medicine at Stanford Health Care in Palo Alto, CA.
Disclosure: The authors report no relevant financial relationships.
References
1. Olsen E, Vonderheid E, Pimpinelli N, et al; ISCL/EORTC. Revisions to the staging and classification of mycosis fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110(6):1713-1722.
2. Vonderheid EC, Pena J, Nowell P. Sézary cell counts in erythrodermic cutaneous T-cell lymphoma: implications for prognosis and staging. Leuk Lymphoma. 2006;47(9):1841-1856.
3. Çetinözman F, Jansen PM, Willemze R. Expression of programmed death-1 in skin biopsies of benign inflammatory vs. lymphomatous erythroderma. Br J Dermatol. 2014;171(3):499-504.
4. Cetinözman F, Jansen PM, Vermeer MH, Willemze R. Differential expression of programmed death-1 (PD-1) in Sézary syndrome and mycosis fungoides. Arch Dermatol. 2012;148(12):1379-1385.
This case is reminiscent of Sézary syndrome as the defining histomorphology in a patient with stage 1B mycosis fungoides: A clue to early peripheral blood involvement.
A 36-year-old woman presented to her primary care physician with pruritic rash localized to her inner thighs of 7 months’ duration. She had a history of multiple miscarriages, and therefore had peripheral blood cytogenetics and extensive autoimmune work-up from 2014 that were negative. Despite applying triamcinolone topical cream and taking hydroxyzine for 1 month, her pruritus worsened and extended to the hands and feet. Overall, the extent of pruritus was seemingly disparate to the degree of cutaneous disease. Physical examination revealed additional sites of involvement including arms, abdomen, and back. Hyperkeratosis was observed on the palms and soles.

Histopathologic Features
Punch biopsies taken from the bilateral arms revealed a predominantly angiocentric atypical lymphocytic infiltrate in the superficial and mid-dermis with minimal epitheliotropism (Figures 1 and 2). There was some degree of permeation of the interstitium superficially with attendant subepidermal fibrosis. The infiltrate was predominated by small- to intermediate-sized lymphocytes showing nuclear contour irregularity including many cells with hyperconvoluted cerebriform nuclei (Figure 3). A few plasma cells were identified as well. There was a concomitant mild epidermal hyperplasia surmounted by an orthohyperkeratotic scale. While the infiltrate was not significantly epidermotropic, there was very focal folliculotropism, whereby intrafollicular collections of cerebriform lymphocyte defining an interfollicular Pautrier microabscess were identified.


Immunohistochemical Findings
Comprehensive phenotypic studies were performed. The lymphocytic infiltrate was extensively highlighted by CD3 and pan T-cell marker CD2. The CD4 to CD8 ratio showed a clear-cut predominance of CD4 T cells over those of the CD8 subset with a CD4 to CD8 ratio exceeding 5:1 (Figures 4 and 5). The lymphocytes were of the alpha beta subset based on the extent of immunoreactivity for ß-F1. Striking and extensive immunoreactivity of the lymphocytes for programmed death 1 (PD-1) was noted (Figure 6). Marked upregulation of TOX amidst the lymphocytes (Figure 7) including strong nuclear expression of TOX amidst angiocentric lymphocytes was observed. There was focal staining for CD25 (20%-30% of the infiltrate) and the CD7 preparation showed a significant decrement in staining with only 20% of the infiltrate exhibiting positivity (Figure 8). There were a few FOXP3 positive staining T cells, but the majority of the cells did not exhibit a regulatory T-cell phenotype. Overall, the combined light microscopic findings and phenotypic profile was highly suggestive of Sézary syndrome.


Peripheral Blood Studies
She subsequently had peripheral blood studies performed. Her studies disclosed a small percentage of abnormal T cells averaging 0.25% of the entire population examined exhibiting expression of CD2, CD3, and CD4 but with loss of CD5 and CD7. The peripheral blood studies were interpreted as being reflective of minimal involvement by the patient’s cutaneous lymphoma.



Discussion
Sézary syndrome is characterized by the triad of erythroderma, lymphadenopathy, and abnormal T cells (Sézary cells) in the peripheral blood, lymph nodes, and skin. The authors of the 2005 World Health Organization-European Organization for Research and Treatment of Cancer classification for this cutaneous lymphoma suggested that there must be an absolute criterion, which is one of established T-cell clonality in the peripheral blood and skin, ideally demonstrating the same T-cell clone in the 2 different anatomic samples.1 The authors proposed 2 additional criteria, of which at least one must be present. The first is both cytomorphologic and quantitative, requiring a count of 1000 or more Sézary cells per square millimeter in the peripheral blood. The second criterion is immunophenotypic; the CD4 to CD8 T-lymphocyte ratio in the peripheral blood must be in excess of 10:1, with a concomitant loss of pan T-cell markers such as CD2, CD3, CD7, and CD5.1,2
The histopathology was not characteristic for mycosis fungoides as it was a predominantly angiocentric atypical lymphocytic infiltrate with minimal epitheliotropism. This particular morphology and phenotype characterized by a superficial perivascular atypical cerebriform CD4+ PD-1+, and CD7-lymphocytic infiltrate is quite characteristic for Sézary syndrome. Unlike mycosis fungoides where one expects to see conspicuous epidermotropism, including Pautrier microabscesses, biopsies in the setting of Sézary syndrome may be minimally epidermotropic. The infiltrate of Sézary syndrome lies largely within the dermis, exhibiting a vasocentric disposition around vessels of the superficial vascular plexus.
However, this patient did not fulfill criteria for Sézary syndrome based on peripheral blood assessment and lack of erythroderma. This patient was seen by a cutaneous lymphoma expert and was diagnosed as having mycosis fungoides stage 1B. One could speculate that the Sézary syndrome-like biopsy findings predicted peripheral blood involvement albeit not of the magnitude to be considered diagnostic of Sézary syndrome. In this regard, the distinctive histology suggesting a diagnosis of Sézary syndrome could also signify a subset of mycosis fungoides patients who have the potential for a more aggressive clinical course specifically as it applies to peripheral blood involvement.
The expression in this case of PD-1 is a critical phenotypic determinant in the setting of Sézary syndrome. PD-1 is an inhibitory member of the CD28/CTLA-4 family. The PD-1 pathway exerts its function through inhibiting TCR-mediated T-cell proliferation and cytokine production. PD-1 has been useful in differentiating Sézary syndrome from erythrodermic inflammatory dermatoses when expressed in CD4+ T cells over CD8+ T cells.3 Biopsies of Sézary syndrome typically show very prominent expression of PD-1 that far exceeds that in mycosis fungoides.4
It has postulated that Sézary syndrome is a neoplasm of central memory T cells and differs from conventional mycosis fungoides in which the cell of origin is held to be a resident effector T cell. Due to the abnormal pattern of PD-1 staining, a reactive based etiology including in the context of a lymphomatoid drug reaction is not favored.
In summary, the biopsies show a PD-1 and CD4 positive lymphomatoid vascular reaction most suggestive of Sézary syndrome but with clinical features of stage 1B mycosis fungoides and not Sézary syndrome. It is possible that this limited cutaneous presentation along minimal blood involvement may correspond with the earliest manifestation of Sézary syndrome. Conversely this distinct histomorphologic and phenotypic pattern could predict mycosis fungoides patients who may develop peripheral blood involvement with the potential for disease progression to Sézary syndrome.
Dr Magro is the director of dermatopathology at Weill Cornell Medicine in New York, NY.
Dr Tsang is a fellow in transfusion medicine at Stanford Health Care in Palo Alto, CA.
Disclosure: The authors report no relevant financial relationships.
References
1. Olsen E, Vonderheid E, Pimpinelli N, et al; ISCL/EORTC. Revisions to the staging and classification of mycosis fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110(6):1713-1722.
2. Vonderheid EC, Pena J, Nowell P. Sézary cell counts in erythrodermic cutaneous T-cell lymphoma: implications for prognosis and staging. Leuk Lymphoma. 2006;47(9):1841-1856.
3. Çetinözman F, Jansen PM, Willemze R. Expression of programmed death-1 in skin biopsies of benign inflammatory vs. lymphomatous erythroderma. Br J Dermatol. 2014;171(3):499-504.
4. Cetinözman F, Jansen PM, Vermeer MH, Willemze R. Differential expression of programmed death-1 (PD-1) in Sézary syndrome and mycosis fungoides. Arch Dermatol. 2012;148(12):1379-1385.
This case is reminiscent of Sézary syndrome as the defining histomorphology in a patient with stage 1B mycosis fungoides: A clue to early peripheral blood involvement.
A 36-year-old woman presented to her primary care physician with pruritic rash localized to her inner thighs of 7 months’ duration. She had a history of multiple miscarriages, and therefore had peripheral blood cytogenetics and extensive autoimmune work-up from 2014 that were negative. Despite applying triamcinolone topical cream and taking hydroxyzine for 1 month, her pruritus worsened and extended to the hands and feet. Overall, the extent of pruritus was seemingly disparate to the degree of cutaneous disease. Physical examination revealed additional sites of involvement including arms, abdomen, and back. Hyperkeratosis was observed on the palms and soles.
Histopathologic Features
Punch biopsies taken from the bilateral arms revealed a predominantly angiocentric atypical lymphocytic infiltrate in the superficial and mid-dermis with minimal epitheliotropism (Figures 1 and 2). There was some degree of permeation of the interstitium superficially with attendant subepidermal fibrosis. The infiltrate was predominated by small- to intermediate-sized lymphocytes showing nuclear contour irregularity including many cells with hyperconvoluted cerebriform nuclei (Figure 3). A few plasma cells were identified as well. There was a concomitant mild epidermal hyperplasia surmounted by an orthohyperkeratotic scale. While the infiltrate was not significantly epidermotropic, there was very focal folliculotropism, whereby intrafollicular collections of cerebriform lymphocyte defining an interfollicular Pautrier microabscess were identified.
Immunohistochemical Findings
Comprehensive phenotypic studies were performed. The lymphocytic infiltrate was extensively highlighted by CD3 and pan T-cell marker CD2. The CD4 to CD8 ratio showed a clear-cut predominance of CD4 T cells over those of the CD8 subset with a CD4 to CD8 ratio exceeding 5:1 (Figures 4 and 5). The lymphocytes were of the alpha beta subset based on the extent of immunoreactivity for ß-F1. Striking and extensive immunoreactivity of the lymphocytes for programmed death 1 (PD-1) was noted (Figure 6). Marked upregulation of TOX amidst the lymphocytes (Figure 7) including strong nuclear expression of TOX amidst angiocentric lymphocytes was observed. There was focal staining for CD25 (20%-30% of the infiltrate) and the CD7 preparation showed a significant decrement in staining with only 20% of the infiltrate exhibiting positivity (Figure 8). There were a few FOXP3 positive staining T cells, but the majority of the cells did not exhibit a regulatory T-cell phenotype. Overall, the combined light microscopic findings and phenotypic profile was highly suggestive of Sézary syndrome.
Peripheral Blood Studies
She subsequently had peripheral blood studies performed. Her studies disclosed a small percentage of abnormal T cells averaging 0.25% of the entire population examined exhibiting expression of CD2, CD3, and CD4 but with loss of CD5 and CD7. The peripheral blood studies were interpreted as being reflective of minimal involvement by the patient’s cutaneous lymphoma.
Discussion
Sézary syndrome is characterized by the triad of erythroderma, lymphadenopathy, and abnormal T cells (Sézary cells) in the peripheral blood, lymph nodes, and skin. The authors of the 2005 World Health Organization-European Organization for Research and Treatment of Cancer classification for this cutaneous lymphoma suggested that there must be an absolute criterion, which is one of established T-cell clonality in the peripheral blood and skin, ideally demonstrating the same T-cell clone in the 2 different anatomic samples.1 The authors proposed 2 additional criteria, of which at least one must be present. The first is both cytomorphologic and quantitative, requiring a count of 1000 or more Sézary cells per square millimeter in the peripheral blood. The second criterion is immunophenotypic; the CD4 to CD8 T-lymphocyte ratio in the peripheral blood must be in excess of 10:1, with a concomitant loss of pan T-cell markers such as CD2, CD3, CD7, and CD5.1,2
The histopathology was not characteristic for mycosis fungoides as it was a predominantly angiocentric atypical lymphocytic infiltrate with minimal epitheliotropism. This particular morphology and phenotype characterized by a superficial perivascular atypical cerebriform CD4+ PD-1+, and CD7-lymphocytic infiltrate is quite characteristic for Sézary syndrome. Unlike mycosis fungoides where one expects to see conspicuous epidermotropism, including Pautrier microabscesses, biopsies in the setting of Sézary syndrome may be minimally epidermotropic. The infiltrate of Sézary syndrome lies largely within the dermis, exhibiting a vasocentric disposition around vessels of the superficial vascular plexus.
However, this patient did not fulfill criteria for Sézary syndrome based on peripheral blood assessment and lack of erythroderma. This patient was seen by a cutaneous lymphoma expert and was diagnosed as having mycosis fungoides stage 1B. One could speculate that the Sézary syndrome-like biopsy findings predicted peripheral blood involvement albeit not of the magnitude to be considered diagnostic of Sézary syndrome. In this regard, the distinctive histology suggesting a diagnosis of Sézary syndrome could also signify a subset of mycosis fungoides patients who have the potential for a more aggressive clinical course specifically as it applies to peripheral blood involvement.
The expression in this case of PD-1 is a critical phenotypic determinant in the setting of Sézary syndrome. PD-1 is an inhibitory member of the CD28/CTLA-4 family. The PD-1 pathway exerts its function through inhibiting TCR-mediated T-cell proliferation and cytokine production. PD-1 has been useful in differentiating Sézary syndrome from erythrodermic inflammatory dermatoses when expressed in CD4+ T cells over CD8+ T cells.3 Biopsies of Sézary syndrome typically show very prominent expression of PD-1 that far exceeds that in mycosis fungoides.4
It has postulated that Sézary syndrome is a neoplasm of central memory T cells and differs from conventional mycosis fungoides in which the cell of origin is held to be a resident effector T cell. Due to the abnormal pattern of PD-1 staining, a reactive based etiology including in the context of a lymphomatoid drug reaction is not favored.
In summary, the biopsies show a PD-1 and CD4 positive lymphomatoid vascular reaction most suggestive of Sézary syndrome but with clinical features of stage 1B mycosis fungoides and not Sézary syndrome. It is possible that this limited cutaneous presentation along minimal blood involvement may correspond with the earliest manifestation of Sézary syndrome. Conversely this distinct histomorphologic and phenotypic pattern could predict mycosis fungoides patients who may develop peripheral blood involvement with the potential for disease progression to Sézary syndrome.
Dr Magro is the director of dermatopathology at Weill Cornell Medicine in New York, NY.
Dr Tsang is a fellow in transfusion medicine at Stanford Health Care in Palo Alto, CA.
Disclosure: The authors report no relevant financial relationships.
References
1. Olsen E, Vonderheid E, Pimpinelli N, et al; ISCL/EORTC. Revisions to the staging and classification of mycosis fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood. 2007;110(6):1713-1722.
2. Vonderheid EC, Pena J, Nowell P. Sézary cell counts in erythrodermic cutaneous T-cell lymphoma: implications for prognosis and staging. Leuk Lymphoma. 2006;47(9):1841-1856.
3. Çetinözman F, Jansen PM, Willemze R. Expression of programmed death-1 in skin biopsies of benign inflammatory vs. lymphomatous erythroderma. Br J Dermatol. 2014;171(3):499-504.
4. Cetinözman F, Jansen PM, Vermeer MH, Willemze R. Differential expression of programmed death-1 (PD-1) in Sézary syndrome and mycosis fungoides. Arch Dermatol. 2012;148(12):1379-1385.













