
An 8-year-old boy was referred for an evaluation of numerous pigmented macules of the skin. The patient and his family had recently immigrated to the United States. According to the patient’s parents, the cutaneous pigmented lesions first became evident at approximately 1 year of age and have since progressively increased in number. Further history provided by the parents indicates the patient has always been small for his age. There are 3 older full siblings, all in good health with normal growth and without significant cutaneous findings. The parents of the patient are of normal stature and in good health.
Dermatologic examination revealed hundreds of 1 mm to 5 mm dark brown macules located on the face, neck, axillae, back, chest and arms. In addition, an epidermal nevus 1.0 cm x 1.8 cm in size was identified on the patient’s right posterior neck and a café au lait patch, 3.0 cm x 2.0 cm, noted on his medial left thigh. Besides the dermatologic findings, physical examination revealed the patient was short for his age with low set ears, hypertelorism, pectus excavatum and teeth that were irregularly spaced with abnormal angles and tooth decay. A biopsy of one of the characteristic macules from the neck was performed.
What is Your Diagnosis?
To learn the answer, go to page 2
{{pagebreak}}
Diagnosis: LEOPARD Syndrome
Based on his clinical features of hundreds of dark macules of the skin (Figures 1 and 2), his short stature and hypertelorism, the diagnosis of LEOPARD syndrome (LS) was made. Punch biopsy was performed to confirm the diagnosis of lentigines (Figure 3). Histological analysis revealed increased melanin pigment and melanocytes within the basal cell layer consistent with lentigo simplex. Pigment incontinence was identified within the superficial dermis.
Figure 1. Numerous brown macules on neck, upper extremities and trunk.
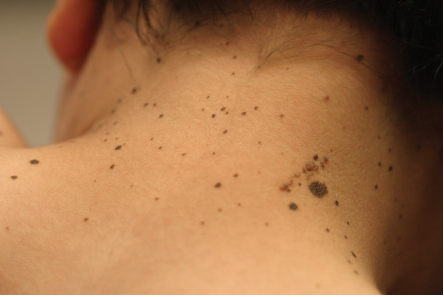
Figure 2. Right posterior neck with tan verrucous plaque, 1.0 cm x 1.8 cm.

Figure 3. 10x hematoxylin and eosin of punch biopsy with increased melanin and melanocytes.
The features of LS were first described in 1936 and the acronym coined in 1969 by Gorlin et al.1,2 It is an autosomal dominant syndrome with high penetrance and variable phenotypes.3 The initial manifestations are frequently cutaneous with the presence of multiple lentigines. However, its clinical features extend beyond cutaneous and its diagnosis is imperative due to the potential for associated cardiac abnormalities.2-5
Clinical Features
LS is a constellation of multiple systemic findings with a prominent cutaneous component. LS is classified as a member of the neuro-cardio-facial- cutaneous group that consists of Noonan syndrome, neurofibromatosis (NF) type 1, Costello syndrome and cardiofaciocutaneous syndrome.2,3
It has been named based on an acronym identifying its cardinal features: lentigines, electrocardiogram (ECG) conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, retardation of growth and sensorineural deafness.1 In 1976, criteria were proposed for diagnosis of LS: multiple lentigines plus 2 of the cardinal features or in the absence of lentigines, 3 of the cardinal features plus a first-degree relative with a diagnosis of LS.4
Today, molecular testing may assist in providing a diagnosis. Its inheritance is autosomal dominant or a sporadic mutation that most frequently is a missense mutation in PTPN11 gene.2,3 In the small percentage of cases that are negative for PTPN11 mutation, RAF1 mutations have been identified.5
Dermatological Features
Typically, the lentigines in LS are usually first identified around the age of 4 or 5 and increase in number until puberty. At that time, the lentigines could number in the thousands. Although lentigines are frequently diffuse, they are usually concentrated on the upper half of the body excluding mucosae.3
Other dermatologic findings that are encountered in LS include café au lait spots, café noir spots and epidermal nevi.3
Other Features
The most concerning anomaly is cardiac involvement, which most frequently presents as ECG conduction abnormalities, hypertrophic cardiomyopathy or pulmonary stenosis.3,6 A review of LS identified ECG irregularities in 75% of cases with ventricular hypertrophy being cited as the most common abnormality.6 Pulmonary stenosis has been the classic structural defect associated with LS. However, recent reviews suggest the incidence of pulmonary stenosis is around 10% to 20%, which is lower than the 40% initially documented.3 It is now believed that hypertrophic cardiomyopathy is more common.3,6 This type of cardiomyopathy most frequently involves the left ventricle and is alarmingly asymptomatic. The onset of the hypertrophic cardiomyopathy is thought to occur before the appearance of lentigines and can progress as lentigines develop. Not all cases of hypertrophic cardiomyopathy lead to outflow obstruction in LS patients. Due to the high incidence of cardiac involvement, complete cardiac evaluation is warranted including ECG and echocardiography.3,6
Hypertelorism is identified in the vast majority of individuals with LS. The most common genital abnormality documented is bilateral cryptorchidism, which is thought to occur in approximately 50% of males.3 In addition, hypospadias and genital hypoplasia are seen.3 A majority of patients with LS have height below the 25th percentile.3,4 The occurrence of sensorineural deafness is reported to be around 15% to 25%.
In addition to the cardinal features, other findings that can be associated with LS include pectus carinatum or excavatum, low set ears, ptosis, abnormal dentition and learning disabilities.3 Additional but less frequent cardiac abnormalities include mitral valve prolapse and septal defects.
Differential Diagnosis
Noonan syndrome and LS often share a number of clinical characteristics: short stature, cryptorchidism, cardiac abnormalities including pulmonary stenosis and hypertrophic cardiomyopathy, short stature, pectus excavatum or carinatum and mutation of the PTPN11 gene.7 However, Noonan syndrome presents with the absence of the hallmark lentigines of LS and a gain of function of tyrosine phosphatase activity.7,8 LS has a decreased tyrosine phosphatase activity.
Carney syndrome has the characteristic lentigines of LS, however, the lentigines in Carney syndrome are known to involve the mucosae.9 The presence of blue nevi and myxomas are also characteristic of Carney syndrome and absent in LS.
NF type 1 should also be considered in the differential diagnosis. It is due to a mutation in the NF1 gene with café au lait macules as the earliest clinical feature.10 These macules often increase in size and number throughout childhood. Axillary freckling and neurofibromas appear in older children and adolescents.10 The Table outlines the characteristics and genetics associated with each differential diagnosis.
 Our Patient
Our Patient
This patient met the criteria for LS as he exhibited multiple lentigines, ocular hypertelorism and growth retardation. Other findings included mild proptosis, a wide triangularly-shaped face, long smooth philtrum and a thin upper lip. The nasal bridge and tip were broad. Both ears were low set and large with upturned earlobes. He also demonstrated superior pectus carinatum with inferior pectus excavatum. In addition to the lentigines, the patient presented with a café au lait patch and epidermal nevus that are commonly identified in LS.
Due to the frequent association of LS with cardiac anomalies, a pediatric cardiology consultation was requested. The patient’s ECG and echocardiogram were read as normal. The young patient was referred to a pediatric geneticist who confirmed the diagnosis of LS. His height was 119.4 cm and his weight was 20.8 kg. Both his height and weight place him less than the fifth percentile in these categories compared to males his age.
We requested the patient to follow-up annually with dermatology to assist with compliance regarding his yearly cardiac evaluations as well as auditory and clinical examinations. The patient’s family was also advised to consult genetic counseling to discuss the possibility of transmission of LS to the patient’s offspring due to its mendelian inheritance.
Conclusion
This young patient presented with many of the classic clinical findings associated with LS. The diagnosis of LS requires genetic counseling for the patient and family. In addition to an ongoing cardiac evaluation, a complete clinical examination with emphasis on the genitourinary and skeletal evaluations should be performed. An age appropriate hearing assessment is also warranted.
Dr. Leigh Sutton is a PGY2 in dermatology at Scott and White in Temple, TX.
Ms. Elizabeth Sutton is a medical student at the University of Nebraska Medical Center in Omaha, NE.
Dr. Margaret Sutton is a board certified dermatologist in private practice in Lincoln, NE.
Disclosure: The authors no relevant financial relationships.
References
1. Sarkozy A, Digilio MC, Dallapiccola B. Leopard syndrome. Orphanet J Rare Dis. 2008;3:13.
2. Gorlin RJ, Anderson RC, Blaw M. Mutiple lentigines syndrome. Am J Dis Child. 1969;117(6):652-662.
3. Kalev I, Muru K, Teek R, et al. LEOPARD syndrome with recurrent PTPN11 mutation Y279C and different cutaneous manifestations: Two case reports and a review of the literature. Eur J Pediatr. 2010;169(4):469-473.
4. Voron DA, Hatfield HH, Kalkhoff RK. Multiple lentigines syndrome. Case report and review of literature. Am J Med. 1976;60(3):447-456.
5. Pandit B, Sarkozy A, Pennacchio LA, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007;39(8):1007-1012.
6. Limongelli G, Pacileo G, Marino B, et al. Prevalence and clinical significance of cardiovascular abnormalities in patients with the LEOPARD syndrome. Am J Cardiol. 2007;100(4):736-741.
7. Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet. 2013;381(9863):333-342.
8. Aoki Y, Niihori T, Banjo T, et al. Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet. 2013;93(1):173-180.
9. Horvath A, Stratakis CA. Carney complex and lentiginosis. Pigment Cell Melanoma Res. 2009;22(5):580-587.
10. Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 2009;61(1):1-14.

An 8-year-old boy was referred for an evaluation of numerous pigmented macules of the skin. The patient and his family had recently immigrated to the United States. According to the patient’s parents, the cutaneous pigmented lesions first became evident at approximately 1 year of age and have since progressively increased in number. Further history provided by the parents indicates the patient has always been small for his age. There are 3 older full siblings, all in good health with normal growth and without significant cutaneous findings. The parents of the patient are of normal stature and in good health.
Dermatologic examination revealed hundreds of 1 mm to 5 mm dark brown macules located on the face, neck, axillae, back, chest and arms. In addition, an epidermal nevus 1.0 cm x 1.8 cm in size was identified on the patient’s right posterior neck and a café au lait patch, 3.0 cm x 2.0 cm, noted on his medial left thigh. Besides the dermatologic findings, physical examination revealed the patient was short for his age with low set ears, hypertelorism, pectus excavatum and teeth that were irregularly spaced with abnormal angles and tooth decay. A biopsy of one of the characteristic macules from the neck was performed.
What is Your Diagnosis?
Diagnosis: LEOPARD Syndrome
Based on his clinical features of hundreds of dark macules of the skin (Figures 1 and 2), his short stature and hypertelorism, the diagnosis of LEOPARD syndrome (LS) was made. Punch biopsy was performed to confirm the diagnosis of lentigines (Figure 3). Histological analysis revealed increased melanin pigment and melanocytes within the basal cell layer consistent with lentigo simplex. Pigment incontinence was identified within the superficial dermis.
Figure 1. Numerous brown macules on neck, upper extremities and trunk.

Figure 2. Right posterior neck with tan verrucous plaque, 1.0 cm x 1.8 cm.

Figure 3. 10x hematoxylin and eosin of punch biopsy with increased melanin and melanocytes.
The features of LS were first described in 1936 and the acronym coined in 1969 by Gorlin et al.1,2 It is an autosomal dominant syndrome with high penetrance and variable phenotypes.3 The initial manifestations are frequently cutaneous with the presence of multiple lentigines. However, its clinical features extend beyond cutaneous and its diagnosis is imperative due to the potential for associated cardiac abnormalities.2-5
Clinical Features
LS is a constellation of multiple systemic findings with a prominent cutaneous component. LS is classified as a member of the neuro-cardio-facial- cutaneous group that consists of Noonan syndrome, neurofibromatosis (NF) type 1, Costello syndrome and cardiofaciocutaneous syndrome.2,3
It has been named based on an acronym identifying its cardinal features: lentigines, electrocardiogram (ECG) conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, retardation of growth and sensorineural deafness.1 In 1976, criteria were proposed for diagnosis of LS: multiple lentigines plus 2 of the cardinal features or in the absence of lentigines, 3 of the cardinal features plus a first-degree relative with a diagnosis of LS.4
Today, molecular testing may assist in providing a diagnosis. Its inheritance is autosomal dominant or a sporadic mutation that most frequently is a missense mutation in PTPN11 gene.2,3 In the small percentage of cases that are negative for PTPN11 mutation, RAF1 mutations have been identified.5
Dermatological Features
Typically, the lentigines in LS are usually first identified around the age of 4 or 5 and increase in number until puberty. At that time, the lentigines could number in the thousands. Although lentigines are frequently diffuse, they are usually concentrated on the upper half of the body excluding mucosae.3
Other dermatologic findings that are encountered in LS include café au lait spots, café noir spots and epidermal nevi.3
Other Features
The most concerning anomaly is cardiac involvement, which most frequently presents as ECG conduction abnormalities, hypertrophic cardiomyopathy or pulmonary stenosis.3,6 A review of LS identified ECG irregularities in 75% of cases with ventricular hypertrophy being cited as the most common abnormality.6 Pulmonary stenosis has been the classic structural defect associated with LS. However, recent reviews suggest the incidence of pulmonary stenosis is around 10% to 20%, which is lower than the 40% initially documented.3 It is now believed that hypertrophic cardiomyopathy is more common.3,6 This type of cardiomyopathy most frequently involves the left ventricle and is alarmingly asymptomatic. The onset of the hypertrophic cardiomyopathy is thought to occur before the appearance of lentigines and can progress as lentigines develop. Not all cases of hypertrophic cardiomyopathy lead to outflow obstruction in LS patients. Due to the high incidence of cardiac involvement, complete cardiac evaluation is warranted including ECG and echocardiography.3,6
Hypertelorism is identified in the vast majority of individuals with LS. The most common genital abnormality documented is bilateral cryptorchidism, which is thought to occur in approximately 50% of males.3 In addition, hypospadias and genital hypoplasia are seen.3 A majority of patients with LS have height below the 25th percentile.3,4 The occurrence of sensorineural deafness is reported to be around 15% to 25%.
In addition to the cardinal features, other findings that can be associated with LS include pectus carinatum or excavatum, low set ears, ptosis, abnormal dentition and learning disabilities.3 Additional but less frequent cardiac abnormalities include mitral valve prolapse and septal defects.
Differential Diagnosis
Noonan syndrome and LS often share a number of clinical characteristics: short stature, cryptorchidism, cardiac abnormalities including pulmonary stenosis and hypertrophic cardiomyopathy, short stature, pectus excavatum or carinatum and mutation of the PTPN11 gene.7 However, Noonan syndrome presents with the absence of the hallmark lentigines of LS and a gain of function of tyrosine phosphatase activity.7,8 LS has a decreased tyrosine phosphatase activity.
Carney syndrome has the characteristic lentigines of LS, however, the lentigines in Carney syndrome are known to involve the mucosae.9 The presence of blue nevi and myxomas are also characteristic of Carney syndrome and absent in LS.
NF type 1 should also be considered in the differential diagnosis. It is due to a mutation in the NF1 gene with café au lait macules as the earliest clinical feature.10 These macules often increase in size and number throughout childhood. Axillary freckling and neurofibromas appear in older children and adolescents.10 The Table outlines the characteristics and genetics associated with each differential diagnosis.
 Our Patient
Our Patient
This patient met the criteria for LS as he exhibited multiple lentigines, ocular hypertelorism and growth retardation. Other findings included mild proptosis, a wide triangularly-shaped face, long smooth philtrum and a thin upper lip. The nasal bridge and tip were broad. Both ears were low set and large with upturned earlobes. He also demonstrated superior pectus carinatum with inferior pectus excavatum. In addition to the lentigines, the patient presented with a café au lait patch and epidermal nevus that are commonly identified in LS.
Due to the frequent association of LS with cardiac anomalies, a pediatric cardiology consultation was requested. The patient’s ECG and echocardiogram were read as normal. The young patient was referred to a pediatric geneticist who confirmed the diagnosis of LS. His height was 119.4 cm and his weight was 20.8 kg. Both his height and weight place him less than the fifth percentile in these categories compared to males his age.
We requested the patient to follow-up annually with dermatology to assist with compliance regarding his yearly cardiac evaluations as well as auditory and clinical examinations. The patient’s family was also advised to consult genetic counseling to discuss the possibility of transmission of LS to the patient’s offspring due to its mendelian inheritance.
Conclusion
This young patient presented with many of the classic clinical findings associated with LS. The diagnosis of LS requires genetic counseling for the patient and family. In addition to an ongoing cardiac evaluation, a complete clinical examination with emphasis on the genitourinary and skeletal evaluations should be performed. An age appropriate hearing assessment is also warranted.
Dr. Leigh Sutton is a PGY2 in dermatology at Scott and White in Temple, TX.
Ms. Elizabeth Sutton is a medical student at the University of Nebraska Medical Center in Omaha, NE.
Dr. Margaret Sutton is a board certified dermatologist in private practice in Lincoln, NE.
Disclosure: The authors no relevant financial relationships.
References
1. Sarkozy A, Digilio MC, Dallapiccola B. Leopard syndrome. Orphanet J Rare Dis. 2008;3:13.
2. Gorlin RJ, Anderson RC, Blaw M. Mutiple lentigines syndrome. Am J Dis Child. 1969;117(6):652-662.
3. Kalev I, Muru K, Teek R, et al. LEOPARD syndrome with recurrent PTPN11 mutation Y279C and different cutaneous manifestations: Two case reports and a review of the literature. Eur J Pediatr. 2010;169(4):469-473.
4. Voron DA, Hatfield HH, Kalkhoff RK. Multiple lentigines syndrome. Case report and review of literature. Am J Med. 1976;60(3):447-456.
5. Pandit B, Sarkozy A, Pennacchio LA, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007;39(8):1007-1012.
6. Limongelli G, Pacileo G, Marino B, et al. Prevalence and clinical significance of cardiovascular abnormalities in patients with the LEOPARD syndrome. Am J Cardiol. 2007;100(4):736-741.
7. Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet. 2013;381(9863):333-342.
8. Aoki Y, Niihori T, Banjo T, et al. Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet. 2013;93(1):173-180.
9. Horvath A, Stratakis CA. Carney complex and lentiginosis. Pigment Cell Melanoma Res. 2009;22(5):580-587.
10. Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 2009;61(1):1-14.
An 8-year-old boy was referred for an evaluation of numerous pigmented macules of the skin. The patient and his family had recently immigrated to the United States. According to the patient’s parents, the cutaneous pigmented lesions first became evident at approximately 1 year of age and have since progressively increased in number. Further history provided by the parents indicates the patient has always been small for his age. There are 3 older full siblings, all in good health with normal growth and without significant cutaneous findings. The parents of the patient are of normal stature and in good health.
Dermatologic examination revealed hundreds of 1 mm to 5 mm dark brown macules located on the face, neck, axillae, back, chest and arms. In addition, an epidermal nevus 1.0 cm x 1.8 cm in size was identified on the patient’s right posterior neck and a café au lait patch, 3.0 cm x 2.0 cm, noted on his medial left thigh. Besides the dermatologic findings, physical examination revealed the patient was short for his age with low set ears, hypertelorism, pectus excavatum and teeth that were irregularly spaced with abnormal angles and tooth decay. A biopsy of one of the characteristic macules from the neck was performed.
What is Your Diagnosis?
,
An 8-year-old boy was referred for an evaluation of numerous pigmented macules of the skin. The patient and his family had recently immigrated to the United States. According to the patient’s parents, the cutaneous pigmented lesions first became evident at approximately 1 year of age and have since progressively increased in number. Further history provided by the parents indicates the patient has always been small for his age. There are 3 older full siblings, all in good health with normal growth and without significant cutaneous findings. The parents of the patient are of normal stature and in good health.
Dermatologic examination revealed hundreds of 1 mm to 5 mm dark brown macules located on the face, neck, axillae, back, chest and arms. In addition, an epidermal nevus 1.0 cm x 1.8 cm in size was identified on the patient’s right posterior neck and a café au lait patch, 3.0 cm x 2.0 cm, noted on his medial left thigh. Besides the dermatologic findings, physical examination revealed the patient was short for his age with low set ears, hypertelorism, pectus excavatum and teeth that were irregularly spaced with abnormal angles and tooth decay. A biopsy of one of the characteristic macules from the neck was performed.
What is Your Diagnosis?
To learn the answer, go to page 2
{{pagebreak}}
Diagnosis: LEOPARD Syndrome
Based on his clinical features of hundreds of dark macules of the skin (Figures 1 and 2), his short stature and hypertelorism, the diagnosis of LEOPARD syndrome (LS) was made. Punch biopsy was performed to confirm the diagnosis of lentigines (Figure 3). Histological analysis revealed increased melanin pigment and melanocytes within the basal cell layer consistent with lentigo simplex. Pigment incontinence was identified within the superficial dermis.
Figure 1. Numerous brown macules on neck, upper extremities and trunk.
Figure 2. Right posterior neck with tan verrucous plaque, 1.0 cm x 1.8 cm.
Figure 3. 10x hematoxylin and eosin of punch biopsy with increased melanin and melanocytes.
The features of LS were first described in 1936 and the acronym coined in 1969 by Gorlin et al.1,2 It is an autosomal dominant syndrome with high penetrance and variable phenotypes.3 The initial manifestations are frequently cutaneous with the presence of multiple lentigines. However, its clinical features extend beyond cutaneous and its diagnosis is imperative due to the potential for associated cardiac abnormalities.2-5
Clinical Features
LS is a constellation of multiple systemic findings with a prominent cutaneous component. LS is classified as a member of the neuro-cardio-facial- cutaneous group that consists of Noonan syndrome, neurofibromatosis (NF) type 1, Costello syndrome and cardiofaciocutaneous syndrome.2,3
It has been named based on an acronym identifying its cardinal features: lentigines, electrocardiogram (ECG) conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, retardation of growth and sensorineural deafness.1 In 1976, criteria were proposed for diagnosis of LS: multiple lentigines plus 2 of the cardinal features or in the absence of lentigines, 3 of the cardinal features plus a first-degree relative with a diagnosis of LS.4
Today, molecular testing may assist in providing a diagnosis. Its inheritance is autosomal dominant or a sporadic mutation that most frequently is a missense mutation in PTPN11 gene.2,3 In the small percentage of cases that are negative for PTPN11 mutation, RAF1 mutations have been identified.5
Dermatological Features
Typically, the lentigines in LS are usually first identified around the age of 4 or 5 and increase in number until puberty. At that time, the lentigines could number in the thousands. Although lentigines are frequently diffuse, they are usually concentrated on the upper half of the body excluding mucosae.3
Other dermatologic findings that are encountered in LS include café au lait spots, café noir spots and epidermal nevi.3
Other Features
The most concerning anomaly is cardiac involvement, which most frequently presents as ECG conduction abnormalities, hypertrophic cardiomyopathy or pulmonary stenosis.3,6 A review of LS identified ECG irregularities in 75% of cases with ventricular hypertrophy being cited as the most common abnormality.6 Pulmonary stenosis has been the classic structural defect associated with LS. However, recent reviews suggest the incidence of pulmonary stenosis is around 10% to 20%, which is lower than the 40% initially documented.3 It is now believed that hypertrophic cardiomyopathy is more common.3,6 This type of cardiomyopathy most frequently involves the left ventricle and is alarmingly asymptomatic. The onset of the hypertrophic cardiomyopathy is thought to occur before the appearance of lentigines and can progress as lentigines develop. Not all cases of hypertrophic cardiomyopathy lead to outflow obstruction in LS patients. Due to the high incidence of cardiac involvement, complete cardiac evaluation is warranted including ECG and echocardiography.3,6
Hypertelorism is identified in the vast majority of individuals with LS. The most common genital abnormality documented is bilateral cryptorchidism, which is thought to occur in approximately 50% of males.3 In addition, hypospadias and genital hypoplasia are seen.3 A majority of patients with LS have height below the 25th percentile.3,4 The occurrence of sensorineural deafness is reported to be around 15% to 25%.
In addition to the cardinal features, other findings that can be associated with LS include pectus carinatum or excavatum, low set ears, ptosis, abnormal dentition and learning disabilities.3 Additional but less frequent cardiac abnormalities include mitral valve prolapse and septal defects.
Differential Diagnosis
Noonan syndrome and LS often share a number of clinical characteristics: short stature, cryptorchidism, cardiac abnormalities including pulmonary stenosis and hypertrophic cardiomyopathy, short stature, pectus excavatum or carinatum and mutation of the PTPN11 gene.7 However, Noonan syndrome presents with the absence of the hallmark lentigines of LS and a gain of function of tyrosine phosphatase activity.7,8 LS has a decreased tyrosine phosphatase activity.
Carney syndrome has the characteristic lentigines of LS, however, the lentigines in Carney syndrome are known to involve the mucosae.9 The presence of blue nevi and myxomas are also characteristic of Carney syndrome and absent in LS.
NF type 1 should also be considered in the differential diagnosis. It is due to a mutation in the NF1 gene with café au lait macules as the earliest clinical feature.10 These macules often increase in size and number throughout childhood. Axillary freckling and neurofibromas appear in older children and adolescents.10 The Table outlines the characteristics and genetics associated with each differential diagnosis.
Our Patient
This patient met the criteria for LS as he exhibited multiple lentigines, ocular hypertelorism and growth retardation. Other findings included mild proptosis, a wide triangularly-shaped face, long smooth philtrum and a thin upper lip. The nasal bridge and tip were broad. Both ears were low set and large with upturned earlobes. He also demonstrated superior pectus carinatum with inferior pectus excavatum. In addition to the lentigines, the patient presented with a café au lait patch and epidermal nevus that are commonly identified in LS.
Due to the frequent association of LS with cardiac anomalies, a pediatric cardiology consultation was requested. The patient’s ECG and echocardiogram were read as normal. The young patient was referred to a pediatric geneticist who confirmed the diagnosis of LS. His height was 119.4 cm and his weight was 20.8 kg. Both his height and weight place him less than the fifth percentile in these categories compared to males his age.
We requested the patient to follow-up annually with dermatology to assist with compliance regarding his yearly cardiac evaluations as well as auditory and clinical examinations. The patient’s family was also advised to consult genetic counseling to discuss the possibility of transmission of LS to the patient’s offspring due to its mendelian inheritance.
Conclusion
This young patient presented with many of the classic clinical findings associated with LS. The diagnosis of LS requires genetic counseling for the patient and family. In addition to an ongoing cardiac evaluation, a complete clinical examination with emphasis on the genitourinary and skeletal evaluations should be performed. An age appropriate hearing assessment is also warranted.
Dr. Leigh Sutton is a PGY2 in dermatology at Scott and White in Temple, TX.
Ms. Elizabeth Sutton is a medical student at the University of Nebraska Medical Center in Omaha, NE.
Dr. Margaret Sutton is a board certified dermatologist in private practice in Lincoln, NE.
Disclosure: The authors no relevant financial relationships.
References
1. Sarkozy A, Digilio MC, Dallapiccola B. Leopard syndrome. Orphanet J Rare Dis. 2008;3:13.
2. Gorlin RJ, Anderson RC, Blaw M. Mutiple lentigines syndrome. Am J Dis Child. 1969;117(6):652-662.
3. Kalev I, Muru K, Teek R, et al. LEOPARD syndrome with recurrent PTPN11 mutation Y279C and different cutaneous manifestations: Two case reports and a review of the literature. Eur J Pediatr. 2010;169(4):469-473.
4. Voron DA, Hatfield HH, Kalkhoff RK. Multiple lentigines syndrome. Case report and review of literature. Am J Med. 1976;60(3):447-456.
5. Pandit B, Sarkozy A, Pennacchio LA, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007;39(8):1007-1012.
6. Limongelli G, Pacileo G, Marino B, et al. Prevalence and clinical significance of cardiovascular abnormalities in patients with the LEOPARD syndrome. Am J Cardiol. 2007;100(4):736-741.
7. Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet. 2013;381(9863):333-342.
8. Aoki Y, Niihori T, Banjo T, et al. Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet. 2013;93(1):173-180.
9. Horvath A, Stratakis CA. Carney complex and lentiginosis. Pigment Cell Melanoma Res. 2009;22(5):580-587.
10. Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 2009;61(1):1-14.
An 8-year-old boy was referred for an evaluation of numerous pigmented macules of the skin. The patient and his family had recently immigrated to the United States. According to the patient’s parents, the cutaneous pigmented lesions first became evident at approximately 1 year of age and have since progressively increased in number. Further history provided by the parents indicates the patient has always been small for his age. There are 3 older full siblings, all in good health with normal growth and without significant cutaneous findings. The parents of the patient are of normal stature and in good health.
Dermatologic examination revealed hundreds of 1 mm to 5 mm dark brown macules located on the face, neck, axillae, back, chest and arms. In addition, an epidermal nevus 1.0 cm x 1.8 cm in size was identified on the patient’s right posterior neck and a café au lait patch, 3.0 cm x 2.0 cm, noted on his medial left thigh. Besides the dermatologic findings, physical examination revealed the patient was short for his age with low set ears, hypertelorism, pectus excavatum and teeth that were irregularly spaced with abnormal angles and tooth decay. A biopsy of one of the characteristic macules from the neck was performed.
What is Your Diagnosis?
Diagnosis: LEOPARD Syndrome
Based on his clinical features of hundreds of dark macules of the skin (Figures 1 and 2), his short stature and hypertelorism, the diagnosis of LEOPARD syndrome (LS) was made. Punch biopsy was performed to confirm the diagnosis of lentigines (Figure 3). Histological analysis revealed increased melanin pigment and melanocytes within the basal cell layer consistent with lentigo simplex. Pigment incontinence was identified within the superficial dermis.
Figure 1. Numerous brown macules on neck, upper extremities and trunk.
Figure 2. Right posterior neck with tan verrucous plaque, 1.0 cm x 1.8 cm.
Figure 3. 10x hematoxylin and eosin of punch biopsy with increased melanin and melanocytes.
The features of LS were first described in 1936 and the acronym coined in 1969 by Gorlin et al.1,2 It is an autosomal dominant syndrome with high penetrance and variable phenotypes.3 The initial manifestations are frequently cutaneous with the presence of multiple lentigines. However, its clinical features extend beyond cutaneous and its diagnosis is imperative due to the potential for associated cardiac abnormalities.2-5
Clinical Features
LS is a constellation of multiple systemic findings with a prominent cutaneous component. LS is classified as a member of the neuro-cardio-facial- cutaneous group that consists of Noonan syndrome, neurofibromatosis (NF) type 1, Costello syndrome and cardiofaciocutaneous syndrome.2,3
It has been named based on an acronym identifying its cardinal features: lentigines, electrocardiogram (ECG) conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, retardation of growth and sensorineural deafness.1 In 1976, criteria were proposed for diagnosis of LS: multiple lentigines plus 2 of the cardinal features or in the absence of lentigines, 3 of the cardinal features plus a first-degree relative with a diagnosis of LS.4
Today, molecular testing may assist in providing a diagnosis. Its inheritance is autosomal dominant or a sporadic mutation that most frequently is a missense mutation in PTPN11 gene.2,3 In the small percentage of cases that are negative for PTPN11 mutation, RAF1 mutations have been identified.5
Dermatological Features
Typically, the lentigines in LS are usually first identified around the age of 4 or 5 and increase in number until puberty. At that time, the lentigines could number in the thousands. Although lentigines are frequently diffuse, they are usually concentrated on the upper half of the body excluding mucosae.3
Other dermatologic findings that are encountered in LS include café au lait spots, café noir spots and epidermal nevi.3
Other Features
The most concerning anomaly is cardiac involvement, which most frequently presents as ECG conduction abnormalities, hypertrophic cardiomyopathy or pulmonary stenosis.3,6 A review of LS identified ECG irregularities in 75% of cases with ventricular hypertrophy being cited as the most common abnormality.6 Pulmonary stenosis has been the classic structural defect associated with LS. However, recent reviews suggest the incidence of pulmonary stenosis is around 10% to 20%, which is lower than the 40% initially documented.3 It is now believed that hypertrophic cardiomyopathy is more common.3,6 This type of cardiomyopathy most frequently involves the left ventricle and is alarmingly asymptomatic. The onset of the hypertrophic cardiomyopathy is thought to occur before the appearance of lentigines and can progress as lentigines develop. Not all cases of hypertrophic cardiomyopathy lead to outflow obstruction in LS patients. Due to the high incidence of cardiac involvement, complete cardiac evaluation is warranted including ECG and echocardiography.3,6
Hypertelorism is identified in the vast majority of individuals with LS. The most common genital abnormality documented is bilateral cryptorchidism, which is thought to occur in approximately 50% of males.3 In addition, hypospadias and genital hypoplasia are seen.3 A majority of patients with LS have height below the 25th percentile.3,4 The occurrence of sensorineural deafness is reported to be around 15% to 25%.
In addition to the cardinal features, other findings that can be associated with LS include pectus carinatum or excavatum, low set ears, ptosis, abnormal dentition and learning disabilities.3 Additional but less frequent cardiac abnormalities include mitral valve prolapse and septal defects.
Differential Diagnosis
Noonan syndrome and LS often share a number of clinical characteristics: short stature, cryptorchidism, cardiac abnormalities including pulmonary stenosis and hypertrophic cardiomyopathy, short stature, pectus excavatum or carinatum and mutation of the PTPN11 gene.7 However, Noonan syndrome presents with the absence of the hallmark lentigines of LS and a gain of function of tyrosine phosphatase activity.7,8 LS has a decreased tyrosine phosphatase activity.
Carney syndrome has the characteristic lentigines of LS, however, the lentigines in Carney syndrome are known to involve the mucosae.9 The presence of blue nevi and myxomas are also characteristic of Carney syndrome and absent in LS.
NF type 1 should also be considered in the differential diagnosis. It is due to a mutation in the NF1 gene with café au lait macules as the earliest clinical feature.10 These macules often increase in size and number throughout childhood. Axillary freckling and neurofibromas appear in older children and adolescents.10 The Table outlines the characteristics and genetics associated with each differential diagnosis.
Our Patient
This patient met the criteria for LS as he exhibited multiple lentigines, ocular hypertelorism and growth retardation. Other findings included mild proptosis, a wide triangularly-shaped face, long smooth philtrum and a thin upper lip. The nasal bridge and tip were broad. Both ears were low set and large with upturned earlobes. He also demonstrated superior pectus carinatum with inferior pectus excavatum. In addition to the lentigines, the patient presented with a café au lait patch and epidermal nevus that are commonly identified in LS.
Due to the frequent association of LS with cardiac anomalies, a pediatric cardiology consultation was requested. The patient’s ECG and echocardiogram were read as normal. The young patient was referred to a pediatric geneticist who confirmed the diagnosis of LS. His height was 119.4 cm and his weight was 20.8 kg. Both his height and weight place him less than the fifth percentile in these categories compared to males his age.
We requested the patient to follow-up annually with dermatology to assist with compliance regarding his yearly cardiac evaluations as well as auditory and clinical examinations. The patient’s family was also advised to consult genetic counseling to discuss the possibility of transmission of LS to the patient’s offspring due to its mendelian inheritance.
Conclusion
This young patient presented with many of the classic clinical findings associated with LS. The diagnosis of LS requires genetic counseling for the patient and family. In addition to an ongoing cardiac evaluation, a complete clinical examination with emphasis on the genitourinary and skeletal evaluations should be performed. An age appropriate hearing assessment is also warranted.
Dr. Leigh Sutton is a PGY2 in dermatology at Scott and White in Temple, TX.
Ms. Elizabeth Sutton is a medical student at the University of Nebraska Medical Center in Omaha, NE.
Dr. Margaret Sutton is a board certified dermatologist in private practice in Lincoln, NE.
Disclosure: The authors no relevant financial relationships.
References
1. Sarkozy A, Digilio MC, Dallapiccola B. Leopard syndrome. Orphanet J Rare Dis. 2008;3:13.
2. Gorlin RJ, Anderson RC, Blaw M. Mutiple lentigines syndrome. Am J Dis Child. 1969;117(6):652-662.
3. Kalev I, Muru K, Teek R, et al. LEOPARD syndrome with recurrent PTPN11 mutation Y279C and different cutaneous manifestations: Two case reports and a review of the literature. Eur J Pediatr. 2010;169(4):469-473.
4. Voron DA, Hatfield HH, Kalkhoff RK. Multiple lentigines syndrome. Case report and review of literature. Am J Med. 1976;60(3):447-456.
5. Pandit B, Sarkozy A, Pennacchio LA, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007;39(8):1007-1012.
6. Limongelli G, Pacileo G, Marino B, et al. Prevalence and clinical significance of cardiovascular abnormalities in patients with the LEOPARD syndrome. Am J Cardiol. 2007;100(4):736-741.
7. Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet. 2013;381(9863):333-342.
8. Aoki Y, Niihori T, Banjo T, et al. Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet. 2013;93(1):173-180.
9. Horvath A, Stratakis CA. Carney complex and lentiginosis. Pigment Cell Melanoma Res. 2009;22(5):580-587.
10. Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 2009;61(1):1-14.
Diagnosis: LEOPARD Syndrome
Based on his clinical features of hundreds of dark macules of the skin (Figures 1 and 2), his short stature and hypertelorism, the diagnosis of LEOPARD syndrome (LS) was made. Punch biopsy was performed to confirm the diagnosis of lentigines (Figure 3). Histological analysis revealed increased melanin pigment and melanocytes within the basal cell layer consistent with lentigo simplex. Pigment incontinence was identified within the superficial dermis.
Figure 1. Numerous brown macules on neck, upper extremities and trunk.
Figure 2. Right posterior neck with tan verrucous plaque, 1.0 cm x 1.8 cm.
Figure 3. 10x hematoxylin and eosin of punch biopsy with increased melanin and melanocytes.
The features of LS were first described in 1936 and the acronym coined in 1969 by Gorlin et al.1,2 It is an autosomal dominant syndrome with high penetrance and variable phenotypes.3 The initial manifestations are frequently cutaneous with the presence of multiple lentigines. However, its clinical features extend beyond cutaneous and its diagnosis is imperative due to the potential for associated cardiac abnormalities.2-5
Clinical Features
LS is a constellation of multiple systemic findings with a prominent cutaneous component. LS is classified as a member of the neuro-cardio-facial- cutaneous group that consists of Noonan syndrome, neurofibromatosis (NF) type 1, Costello syndrome and cardiofaciocutaneous syndrome.2,3
It has been named based on an acronym identifying its cardinal features: lentigines, electrocardiogram (ECG) conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, retardation of growth and sensorineural deafness.1 In 1976, criteria were proposed for diagnosis of LS: multiple lentigines plus 2 of the cardinal features or in the absence of lentigines, 3 of the cardinal features plus a first-degree relative with a diagnosis of LS.4
Today, molecular testing may assist in providing a diagnosis. Its inheritance is autosomal dominant or a sporadic mutation that most frequently is a missense mutation in PTPN11 gene.2,3 In the small percentage of cases that are negative for PTPN11 mutation, RAF1 mutations have been identified.5
Dermatological Features
Typically, the lentigines in LS are usually first identified around the age of 4 or 5 and increase in number until puberty. At that time, the lentigines could number in the thousands. Although lentigines are frequently diffuse, they are usually concentrated on the upper half of the body excluding mucosae.3
Other dermatologic findings that are encountered in LS include café au lait spots, café noir spots and epidermal nevi.3
Other Features
The most concerning anomaly is cardiac involvement, which most frequently presents as ECG conduction abnormalities, hypertrophic cardiomyopathy or pulmonary stenosis.3,6 A review of LS identified ECG irregularities in 75% of cases with ventricular hypertrophy being cited as the most common abnormality.6 Pulmonary stenosis has been the classic structural defect associated with LS. However, recent reviews suggest the incidence of pulmonary stenosis is around 10% to 20%, which is lower than the 40% initially documented.3 It is now believed that hypertrophic cardiomyopathy is more common.3,6 This type of cardiomyopathy most frequently involves the left ventricle and is alarmingly asymptomatic. The onset of the hypertrophic cardiomyopathy is thought to occur before the appearance of lentigines and can progress as lentigines develop. Not all cases of hypertrophic cardiomyopathy lead to outflow obstruction in LS patients. Due to the high incidence of cardiac involvement, complete cardiac evaluation is warranted including ECG and echocardiography.3,6
Hypertelorism is identified in the vast majority of individuals with LS. The most common genital abnormality documented is bilateral cryptorchidism, which is thought to occur in approximately 50% of males.3 In addition, hypospadias and genital hypoplasia are seen.3 A majority of patients with LS have height below the 25th percentile.3,4 The occurrence of sensorineural deafness is reported to be around 15% to 25%.
In addition to the cardinal features, other findings that can be associated with LS include pectus carinatum or excavatum, low set ears, ptosis, abnormal dentition and learning disabilities.3 Additional but less frequent cardiac abnormalities include mitral valve prolapse and septal defects.
Differential Diagnosis
Noonan syndrome and LS often share a number of clinical characteristics: short stature, cryptorchidism, cardiac abnormalities including pulmonary stenosis and hypertrophic cardiomyopathy, short stature, pectus excavatum or carinatum and mutation of the PTPN11 gene.7 However, Noonan syndrome presents with the absence of the hallmark lentigines of LS and a gain of function of tyrosine phosphatase activity.7,8 LS has a decreased tyrosine phosphatase activity.
Carney syndrome has the characteristic lentigines of LS, however, the lentigines in Carney syndrome are known to involve the mucosae.9 The presence of blue nevi and myxomas are also characteristic of Carney syndrome and absent in LS.
NF type 1 should also be considered in the differential diagnosis. It is due to a mutation in the NF1 gene with café au lait macules as the earliest clinical feature.10 These macules often increase in size and number throughout childhood. Axillary freckling and neurofibromas appear in older children and adolescents.10 The Table outlines the characteristics and genetics associated with each differential diagnosis.
Our Patient
This patient met the criteria for LS as he exhibited multiple lentigines, ocular hypertelorism and growth retardation. Other findings included mild proptosis, a wide triangularly-shaped face, long smooth philtrum and a thin upper lip. The nasal bridge and tip were broad. Both ears were low set and large with upturned earlobes. He also demonstrated superior pectus carinatum with inferior pectus excavatum. In addition to the lentigines, the patient presented with a café au lait patch and epidermal nevus that are commonly identified in LS.
Due to the frequent association of LS with cardiac anomalies, a pediatric cardiology consultation was requested. The patient’s ECG and echocardiogram were read as normal. The young patient was referred to a pediatric geneticist who confirmed the diagnosis of LS. His height was 119.4 cm and his weight was 20.8 kg. Both his height and weight place him less than the fifth percentile in these categories compared to males his age.
We requested the patient to follow-up annually with dermatology to assist with compliance regarding his yearly cardiac evaluations as well as auditory and clinical examinations. The patient’s family was also advised to consult genetic counseling to discuss the possibility of transmission of LS to the patient’s offspring due to its mendelian inheritance.
Conclusion
This young patient presented with many of the classic clinical findings associated with LS. The diagnosis of LS requires genetic counseling for the patient and family. In addition to an ongoing cardiac evaluation, a complete clinical examination with emphasis on the genitourinary and skeletal evaluations should be performed. An age appropriate hearing assessment is also warranted.
Dr. Leigh Sutton is a PGY2 in dermatology at Scott and White in Temple, TX.
Ms. Elizabeth Sutton is a medical student at the University of Nebraska Medical Center in Omaha, NE.
Dr. Margaret Sutton is a board certified dermatologist in private practice in Lincoln, NE.
Disclosure: The authors no relevant financial relationships.
References
1. Sarkozy A, Digilio MC, Dallapiccola B. Leopard syndrome. Orphanet J Rare Dis. 2008;3:13.
2. Gorlin RJ, Anderson RC, Blaw M. Mutiple lentigines syndrome. Am J Dis Child. 1969;117(6):652-662.
3. Kalev I, Muru K, Teek R, et al. LEOPARD syndrome with recurrent PTPN11 mutation Y279C and different cutaneous manifestations: Two case reports and a review of the literature. Eur J Pediatr. 2010;169(4):469-473.
4. Voron DA, Hatfield HH, Kalkhoff RK. Multiple lentigines syndrome. Case report and review of literature. Am J Med. 1976;60(3):447-456.
5. Pandit B, Sarkozy A, Pennacchio LA, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007;39(8):1007-1012.
6. Limongelli G, Pacileo G, Marino B, et al. Prevalence and clinical significance of cardiovascular abnormalities in patients with the LEOPARD syndrome. Am J Cardiol. 2007;100(4):736-741.
7. Roberts AE, Allanson JE, Tartaglia M, Gelb BD. Noonan syndrome. Lancet. 2013;381(9863):333-342.
8. Aoki Y, Niihori T, Banjo T, et al. Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet. 2013;93(1):173-180.
9. Horvath A, Stratakis CA. Carney complex and lentiginosis. Pigment Cell Melanoma Res. 2009;22(5):580-587.
10. Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 2009;61(1):1-14.

















