

What You Should Know About New Antirheumatic Medications
Rheumatoid arthritis (RA) is a systemic inflammatory polyarthritis that involves small and large joints, and affects approximately 1 percent of the population in the United States.1 The natural progression of the disease leads to irreversible deformity in the hands and feet with destruction of bone and articular cartilage. This may ultimately lead to the loss of function of the extremity. There are numerous extraarticular manifestations of RA (i.e., including vasculitis). They can affect any organ system and result in premature death. Over the last decade, there has been tremendous progress leading to improved understanding of the pathophysiology in RA. The inflammatory cytokines, tumor necrosis factor-a (TNF-a), interleukin-1 (IL-1) and interleukin-6 (IL-6), have been identified as key mediators in the inflammatory process of RA.2 The recent advancements in identifying the inflammatory pathways of RA have led to new and improved treatment strategies that specifically target the inflammatory cytokines. Clinically it has become possible to slow the disease process associated with RA. Rheumatoid arthritis represents an imbalance of the soluble and cellular mediators of inflammation, resulting in a chronic inflammatory process that allows proliferation of synovial tissue (pannus) around articular cartilage and bone.2 The periarticular synovial membrane is characterized by hyperplasia, increased vascularity and an infiltrate of inflammatory cells. It is theorized that auto-antigens presented to CD4+ T lymphocytes illicit a broad immune response. The activated CD4+T lymphocytes stimulate macrophages, monocytes and fibroblasts to produce the cytokines IL-1, IL-6, and TNF-a.2 The cytokines and activated inflammatory cells cause expression of adhesion molecules and angiogenesis of the periarticular synovium. The adhesion molecules allow binding of inflammatory cells and facilitate their entry into the joint. Activated CD4+T lymphocytes are also thought to stimulate osteoclast production that leads to bone resorption. B cells and plasma cells in the synovium produce rheumatoid factor, an IgM antibody to IgG that activates the complement cascade and initiates cellular chemotaxis. Several cytokines involved in the pathologic process of RA have been identified in recent studies. TNF-a and IL-1 have been identified as two key intermediaries of inflammation in RA.2,3 TNF-a is released mainly by monocytes and macrophages, and leads to a pro-inflammatory response in a number of different cell types. TNF-a increases the production of IL-1, IL-6 and IL-8, amplifying the inflammatory response. TNF-a and IL-1 also stimulate fibroblasts, osteoclasts and chondrocytes to release matrix metalloproteinases. This leads to cartilage and tissue destruction. TNF-a stimulates the expression of adhesion molecules on the surface of fibroblasts, allowing the migration of leukocytes into areas of inflammation. IL-1 is produced by monocytes, macrophages, B-cells and activated T-cells in the affected joint. IL-1 also stimulates the release of matrix metalloproteinases in the periarticular synovium from fibroblasts and chondrocytes, resulting in further periarticular destruction.2
How To Diagnose Rheumatoid Arthritis
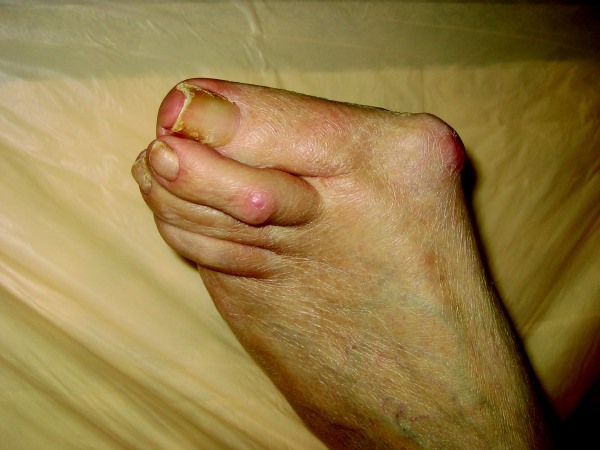
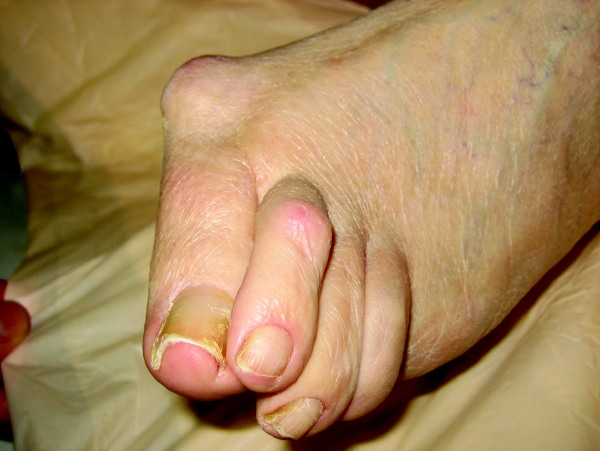
Joint damage begins in the early stages of RA so early detection and diagnosis of RA is essential in the clinical management of the disease.4 RA is a symmetric polyarticular arthritis that frequently affects the small joints of the hands, wrists and feet but can involve any synovial joint. Upon the clinical examination, one may note joints that are swollen, warm and tender. There may also be a local effusion surrounding the joint. Prolonged morning stiffness lasting more than one hour is a hallmark feature of RA. Patients with RA may have large rheumatoid nodules in the lungs, pericardium, myocardium, the palmar surface of the hands and the plantar surface of the foot.5 Chronic RA progresses towards joint destruction and deformity. Radiographic evaluation of joints affected by RA shows erosion of bone and cartilage. Tendons that are anatomically located near affected joints can become inflamed and result in tenosynovitis. This can lead to tendon deformation and subsequent deformity of the affected extremity. Ulnar deviation at the metacarpophalangeal joints and fibular deviation at the metatarsophalangeal joints cause flexion/extension contractures of the distal and proximal interphalangeal joints. This is the characteristic swan-neck or boutonniere deformity. Vasculitis associated with RA may result in ulceration, mononeurtitis and intestinal infarction. There are numerous serologic tests one can employ to diagnose or monitor a patient being treated for RA.5 An examination of joint fluid will show an inflammatory infiltrate of > 10,000 WBC with polymorphonuclear leukocytes being > 80 percent of the observed cells. The presence of C-reactive protein and rheumatoid factor is not necessary for diagnosis but one will see this combination in approximately 80 to 90 percent of patients with RA. Antibodies to cyclic citrullinated peptides (CCPs) have a similar sensitivity as rheumatoid factor but appear to have a high specificity (90 to 98 percent) and may be useful in the early diagnosis of RA. Antinuclear antibodies have been shown to be minimally helpful in diagnosing RA.
Where Do DMARDs Fit In The Armamentarium For RA?
When it comes to managing RA, clinicians seek to provide adequate pain control, maintain function, prevent articular destruction/deformity and possibly achieve remission of the disease.4 Standard nonsteroidal antiinflammatory drug (NSAID) therapy, such as aspirin, ibuprofen, indomethicin or COX-2 inhibitors, are useful in pain control but they do not alter the underlying pathology of RA. They inhibit the inflammatory response by reducing the proinflammatory prostaglandin synthesis mediated by the cyclooxygenase enzyme. NSAID therapy has potential side effects, such as gastrointestinal toxicity leading to ulceration, gastrointestinal bleeding and inhibition of platlet function, so close monitoring of the patient on NSAID therapy is required. Disease-modifying antirheumatic drugs (DMARDs) act to slow or stop the progression of RA via an immunomodulatory response.4 Using these medications early in the course of the disease can slow the articular destruction frequently caused by RA. There are a number of DMARDs used to treat RA. Some of the more common medications include leflunomide (Arava™), methotrexate, sulfasalazine, cyclosporine, hydroxychloroquine and gold salts. Keep in mind, however, that it may be several months before one notes the therapeutic benefit of DMARDs due to the slow onset of action. Clinicians often employ corticosteriod therapy for short periods until the DMARDs achieve a therapeutic response. Monitoring of DMARD effectiveness and safety includes appropriate laboratory tests as well as serial radiographic evaluation for a reduction in articular destruction and osseous erosion. One may consider combination therapy with multiple DMARD medications for patients who show limited disease regression with monotherapy. Side effects associated with the various DMARD medications include but are not limited to hematologic, pulmonary and hepatic side effects as well as possible teratogenicity.
Assessing The Potential Of Etanercept
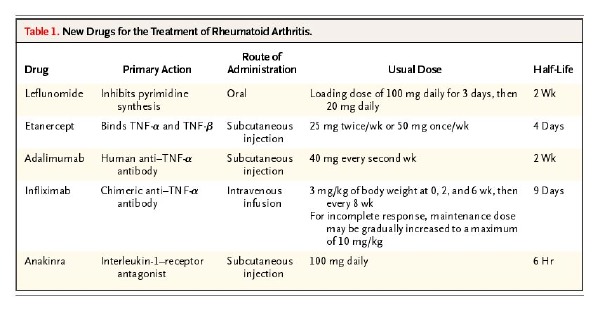
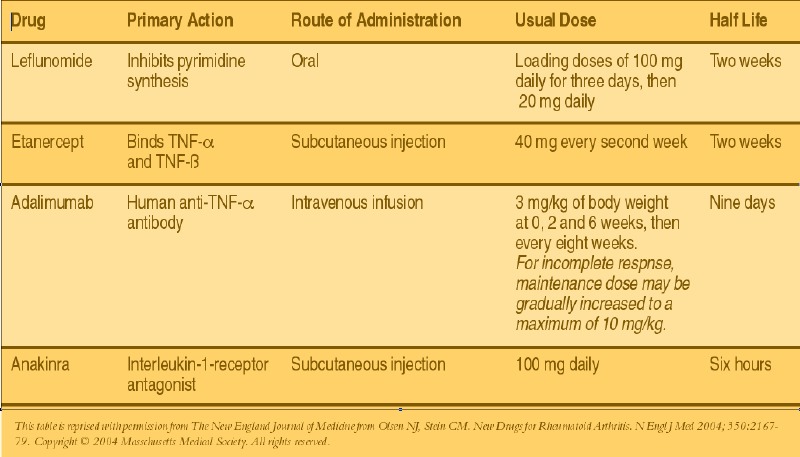
The identification of TNF-a as a key cytokine in the inflammatory cascade has lead to new and novel treatment modalities in the management of RA. There are elevated levels of TNF-a in the synovial pannus in RA patients.2 TNF-a directly binds to TNF receptors on the cellular surface of macrophages, monocytes, T-cells, synovial fibroblasts, endothelial cells and osteoblasts. Binding of TNF-a to its appropriate receptor initiates a pro-inflammatory response. Immunobiologic therapies directly neutralize the pro-inflammatory effects of TNF-a at the cellular level. Inhibiting the effect of TNF-a can significantly slow or even cease the disease progression in addition to improving clinical symptoms in RA. Etanercept (Enbrel™) is a soluble TNF-receptor fusion protein that is linked to the Fc portion of human IgG1.1,6 This protein binds directly to TNF-a and TNF-ß receptors on the cellular surface, preventing signal transduction and commencement of the inflammatory cascade. One can administer etanercept via a subcutaneous injection at 25 mg twice a week or 50 mg once a week with a peak concentration at 50 hours and an approximate half-life of four days. Data from two placebo-controlled trials of 168 and 234 RA patients, who received injections of 25 mg of etanercept twice a week, showed a 50 percent reduction in the number of swollen joints after six months of therapy.7,8 Additionally, synovial biopsies demonstrated a significant decrease in the number of T-cells and plasma cells; reduction in the expression of interleukin-1; and a decline in vascular-cell adhesion molecule 1(VCAM-1) after one month of therapy.9 For patients who have been unresponsive to standard DMARD therapy, researchers have combined etanercept with methotrexate, which facilitated a decrease in clinical symptoms of RA. In a clinical trial comparing etanercept versus methotrexate, there was less radiographic progression among patients who received etanercept.7
What You Should Know About Infliximab
Infliximab (Remicade™) is a chimeric IgG1, anti-TNF-a antibody that binds to soluble- and membrane-bound TNF-a, impairing the binding of TNF-a to its appropriate receptor. Infliximab is unique as it may cause cell lysis of any cell that expresses TNF-a via antibody-dependent and complement-dependent cytotoxicity.2,6 One can administer infliximab intravenously at a dose of 3 mg/kg of body weight at zero, two and six weeks. Clinicans can subsequently prescribe the drug every four to eight weeks for maintenance therapy. Pharmacokinetic data showed that peak concentrations of the drug are patient dependent with an average half-life of nine days. Data from a double-blind, placebo-controlled trial of 73 patients with RA indicated that a single intravenous dose of 10 mg of infliximab per kg resulted in clinical improvement within one week of starting therapy. There was also a significant reduction in the number of swollen joints and reduction in the concentration of C-reactive protein.10 Patients who receive repeated doses of infliximab have developed autoantibodies to infliximab. The clinical importance of infliximab autoantibodies has not been fully established. Researchers have shown that concomitant administration of methotrexate with infliximab may reduce the incidence of autoantibodies against infliximab.10 Radiographic data indicates the prevention of progression in RA patients receiving infliximab and methotrexate.11
Can Adalimumab And Anakinra Have An Impact?
Adalimumab (Humira™) is a recombinant human IgG1 monoclonal antibody that acts similarly to infliximab. Adalimumab binds to human TNF-a, preventing its attachment to the TNF-a receptor and causing lysis of cells expressing TNF-a on their surface. Clinicians can administer adalimumab via subcutaneous injection at a dose of 40 mg every two weeks or every week in more severe patients. Peak concentrations of the drug are achieved at 130 hours after injection with a half-life of two weeks. Researchers have shown that adalimumab is effective compared to placebo in reducing symptoms of joint tenderness and swelling. These effects may be additive when one combines adalimumab with methotrexate.1 Interleukin-1 is a cytokine produced by macrophages, monocytes and other inflammatory cells in the periarticular synovium. IL-1 has pro-inflammatory effects as it stimulates the production of IL-6 and the cyclooxygensase-2 enzyme.12 IL-1 is naturally down-regulated by a IL-1-receptor antagonist, which is thought to be deficient in patients with RA. This allows a continuous inflammatory response. Anakinra (Kineret™) is a recombinant human IL-1-receptor-antagonist that binds to IL-1 receptors. Through competitive inhibition, anakinra suppresses the inflammatory responses caused by IL-1.1 Anakinra may be beneficial to patients who have not responded to previous standard DMARD therapy or TNF-a antagonist therapy. Anakinra has a short half-life of six hours. Therefore, one may administer this medication as a daily subcutaneous injection of 100 mg daily. Researchers have shown that anakinra is effective as monotherapy or in combination with methotrexate, and appears to slow radiographic progression of articular damage.13
Emphasizing Awareness Of Possible Side Effects
One should closely monitor patients receiving TNF-a and IL-1-receptor antagonist therapy. Local skin irritation consisting of minor erythema and itching at the site of injection/infusion is commonly reported among patients receiving infliximab and etanercept.6 The irritation is most common during the first month of therapy and diminishes with subsequent therapy. Local treatment with topical corticosteroids and/or oral/topical antihistamines provides relief of symptoms. Patients receiving TNF-antagonists are at an increased risk of infection. Whenever you consider the use of TNF-antagonist therapy, ensure the patient is screened for latent tuberculosis infection via PPD testing and possibly a chest X-ray. If these patients test positive for this infection, one should obviously pursue appropriate treatment prior to starting TNF-antagonist therapy. Any active infection is a relative contraindication to starting therapy. Atypical mycobacterial infection, aspergillosis, histoplasmosis, coccidioidomycosis, listeriosis, Pneumocystis carinii pneumonia, cryptococcal infections and cytomegalovirus infection have all been reported with use of TNF antagonists.1 Clinicians should obtain baseline and interval complete blood counts (CBC) to monitor for bone marrow suppression although this is unusual. It is uncertain whether TNF antagonists increase the risk of lymphoma. An increase in the incidence of lymphoma has been reported in patients with RA. Concomitant treatment with TNF antagonists may further increase the incidence of lymphoma. Patients with a previous history of demyelinating syndromes (multiple sclerosis) or other autoimmune disorders should probably avoid TNF antagonist therapy since some multiple sclerosis patients have had disease exacerbation when they were treated with these agents. Development of a rash on the hands, forearms and face warrants the discontinuation of therapy. Some patients receiving TNF antagonist therapy developed antinuclear and anti-double stranded DNA antibodies that can be associated with a rare drug-induced lupus syndrome.1
In Conclusion
Although the exact etiology of RA is unknown, a better understanding of the pathophysiology of the disease has led to new therapies. TNF-antagonist and IL-1-receptor-antagonist therapy are effective in the management of RA even though they do not cure the disease. These novel therapies slow the osseous and articular erosion that frequently result in deformity and disability. Early initiation of aggressive therapy can prevent joint destruction and improve short-term and long-term complications associated with RA. With continued laboratory and clinical research, the use of anti-cytokine therapy will likely occupy a larger role in the treatment of not only RA but other chronic inflammatory conditions as well. Dr. Blume is a Clinical Assistant Professor in the Department of Orthopaedics and Rehabilitation at the Yale University School of Medicine. He is also a Fellow of the American College of Foot and Ankle Surgeons, and is the Director of Limb Preservation at the Yale New Haven Hospital in New Haven, Conn. Dr. Cornell is a second-year resident within the Yale/VA Connecticut Healthcare System Podiatric Surgical Residency. Dr. Schoen is an Assistant Clinical Professor of Medicine at Yale University School of Medicine.
References:
1. Olsen NJ, Stein CM. New Drugs for Rheumatoid Arthritis. N Engl J Med 2004; 350:2167-79.
2. Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med 2001; 344:907-16.
3. Houssiau FA. Cytokines in rheumatoid arthritis. Clin Rheumatol 1995; 14:Suppl 2:10-3.
4. O'Dell JR. Therapeutic strategies for rheumatoid arthritis. N Engl J Med 2004; 350:2591-602.
5. Harris ED Jr: Clinical features of rheumatoid arthritis. In Kelley WN, Harris ED Jr, Ruddy s, et al: Textbook of Rheumatology, 5th ed. Philadelphia: WB Saunders, 1997.
6. Corinne RM, Dougados M. Tumor necrosis factor-a blockers in rheumatoid arthritis. BioDrugs 2001; 15(4):251-59.
7. Moreland LW, Schiff MH, Baumgartner SW, et al. Etanercept therapy in rheumatoid arthritis: a randomized controlled trial. Ann Intern Med 1999; 130:478-86.
8. Moreland LW, Baumgartner SW, Schiff MH, et al. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J Med 1997; 337:141-7.
9. Verschueren PD, Markusse H, Smeets TJM et al. Reduced cellularity and expression of adhesion molecules and cytokines after treatment with soluble human recombinant TNF receptor (p75) in RA patients. Arthritis Rheum 1999; 42:Suppl: S401. Abstract.
10. Lipsky PE, Desiree MFM, Heijde VJ, et al. Infliximab and methotrexate in the treatment of rheumatoid arthritis. New Engl J Med; 343:22:1594-1602.
11. Elliot MJ, Maini RN, Feldmann M, et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumor necrosis factor a (cA2) versus placebo in rheumatoid arthritis. Lancet 1994; 344; 1105-10.
12. Maini R, St Clair EW, Breedveld F, et al. Infliximab (chimeric anti-tumor necrosis factor a monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomized phase III trial. Lancet 1999; 354:1932-9.
13. Dianarello CA. Biologic basis for interleukin-1 in disease. Blood 1996; 87:2095-147.
14. Jiang Y, Genant HK, Watt I et al. A multicenter, double-blind, dose-ranging, randomized, placebo-controlled study of recombinant human interleukin-1 receptor antagonist in patients with rheumatoid arthritis: radologic progression and correlation of Genant and Larsen scores. Arthritis Rheum 2000; 43:1001-9.



{kind=link}
{kind=link}
{kind=link}
{kind=link}