


Geriatric Pharmacotherapy Update: New Medications, Recent Releases, and Coming Attractions
INTRODUCTION
Each year, many new medications or new uses for old medications are approved by the U.S. Food and Drug Administration (FDA). In 2004, 119 new medications and 147 new or expanded uses for already approved medications were approved for use in the United States.1 This article will focus on several of these new therapies submitted to the FDA for approval in 2004-2005, with an emphasis on agents potentially useful in conditions experienced by older adults. These agents include trospium (approved 5/28/04), eszopiclone (approved 12/15/04), inhaled insulin (approved 1/27/06), pramlintide (approved 3/16/05), exenatide (approved 4/28/05), and insulin detemir (approved 6/16/05). The mechanism of action (MOA), therapeutic application, side effects, and monitoring of these new drugs will be examined, with some discussion of how they compare to other therapies. Available data will be presented that support conclusive recommendations for use in older adults; however, cautious use is always recommended in this unique population.
URINARY INCONTINENCE
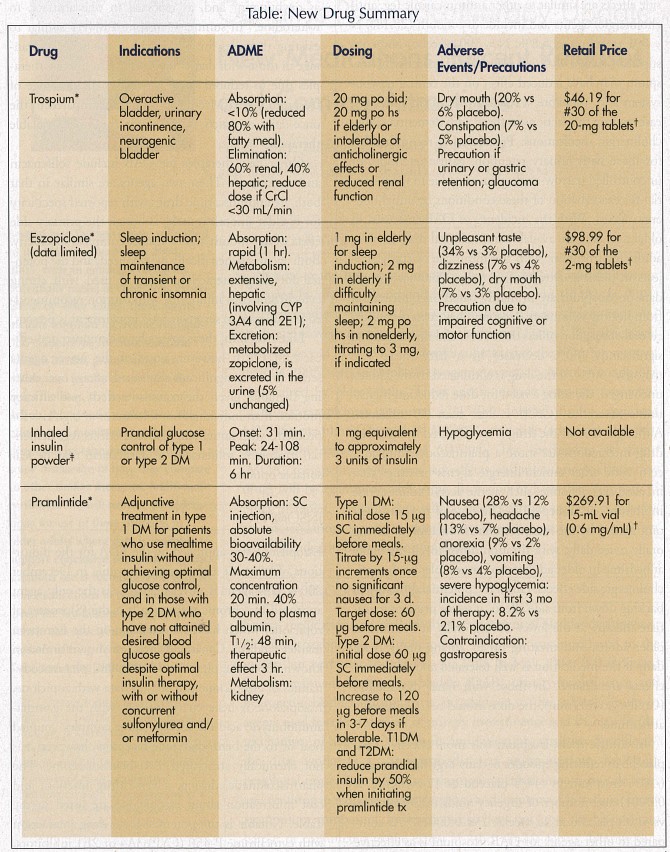
Several medications are available for the treatment of bladder dysfunction, including oxybutynin and tolterodine. Trospium was introduced to the market last year after approval by the FDA in May 2004. It is indicated for the treatment of overactive bladder (OAB), urinary incontinence, and neurogenic bladder. Trospium is an addition to the existing group of agents used for the treatment of urinary incontinence and shares a similar mechanism of action related to its antimuscarinic qualities. Specifically, the drug works as an antispasmodic and antimuscarinic, inhibiting peripheral muscarinic receptors, relaxing urogenital smooth muscle tone, and resulting in reduced urinary urgency and frequency.2 It is a quaternary ammonium compound, however, and so does not cross the blood-brain barrier or conjunctiva to the extent of other similar agents. Despite this, side effects are similar to other antimuscarinic (eg, anticholinergic) agents and include dry mouth (20.1%, N = 591 trospium vs 5.8%, N = 590 placebo) and constipation (9.6% vs 4.6% placebo).3 Theoretically, trospium may have reduced effect on the central nervous system and, therefore, a reduced risk of potential disease or drug interactions in those with dementia taking cholinergic medications. Precaution is recommended for those with urinary retention, gastric retention, or uncontrolled narrow-angle glaucoma due to the potential for exacerbation of these conditions by anticholinergic agents. With the incidence of OAB increased in older age, data are available from clinical trials in older adults for trospium, with the mean age of study subjects in published trials of 63 years. Pharmacokinetic data on trospium are primarily based on information from healthy volunteers. Trospium has extremely limited oral absorption of less than 10%, and this is reduced significantly (80% decrease) by a fatty meal. The majority (60%) of the drug is eliminated by the kidney unchanged, dictating a need for dose reduction if creatinine clearance (CrCl) is less than 30 mL/min. Although 40% of the drug is metabolized by the liver, drug interactions are more a pharmacodynamic concern with other anticholinergic agents or drugs that increase acetylcholine, such as the acetylcholinesterase inhibitors (eg, donepezil).2 The dose of trospium recommended by the manufacturer is initiation at 20 mg orally twice daily, with titration down to 20 mg orally at bedtime in older adults who cannot tolerate the anticholinergic side effects. Given the potential difficulty in backing down from side effects, it seems prudent to initiate therapy at the lower 20 mg orally at bedtime in older adults, and titrating up to 20 mg orally twice daily if the medication is well tolerated and additional effects are desired. In those with renal dysfunction (CrCl < 30 mL/min), the dose should be 20-mg orally at bedtime.
In clinical trials, trospium was more effective than placebo at reducing episodes of daily urge incontinence (-59% trospium vs -44% placebo at 12 weeks; P ≤ 0.0001) and number of urgency voids (-2.3 trospium vs -1.08 placebo at 12 weeks; P ≤ 0.0001).4 As compared to other agents for OAB, trospium is as effective as oxybutynin5 and, as reported in one abstract, to tolterodine.6 In summary, trospium is very similar to existing therapies for OAB, with a theoretical advantage in those with dementia or using cholinergic therapies due to reduced CNS activity. A disadvantage of trospium is the twice-daily dosing as compared to the once-daily or patch formulations for other available therapies.
Other new therapies for OAB include solifenacin and darifenacin. These two agents are similar in that both are antimuscarinic drugs with reported specificity for bladder smooth muscle, although the primary side effect of dry mouth argues against complete specificity. Both agents are hepatically metabolized, and the potential for drug interactions exists if used with strong cytochrome 3A4 inhibitors, with caution recommended if moderate to severe hepatic impairment is present.
Like trospium, these agents have demonstrated efficacy for OAB; however, none of the newer agents seems to offer a significant clinical advantage over existing therapies given the cost, side-effect, and efficacy profiles. If an individual is intolerant or at high risk for problems with anticholinergic side effects (eg, dementia), trospium, solifenacin, or darifenacin may be reasonable options.
INSOMNIA
Eszopiclone was approved by the FDA for the indications of transient and chronic insomnia in December 2004. This agent is novel in that it is the only agent approved for chronic insomnia. It is the (S) isomer of zopiclone, which has been available on the European market and in Canada. Much of the information known about the pharmacokinetics and pharmacodynamics of eszopiclone is based on data with zopiclone. Eszopiclone is believed to interact with the gamma-aminobutyric acid (GABA)-receptor complex coupled near or to the benzodiazepine receptors; however, it is not chemically structured as a benzodiazepine. The pharmacokinetic, dosing, adverse drug reaction, and cost information about eszopiclone are listed in the Table. Notable is the potential for a drug interaction with cytochrome P-450 (CYP) 3A4 or 2E1 inhibitors, leading to excessive eszopiclone levels and the unpleasant taste (dysgeusia) reported by 34% of study subjects as compared to 3% of placebo-treated subjects. In clinical trials, eszopiclone was demonstrated to be more effective than placebo, with significant reduction in wake-time after sleep onset (P < 0.0001) and improved sleep duration (322.3 73.8 min at 1 wk vs 373.5 85.7 min placebo; P < 0.0001).7 These statistically significant differences were maintained at 6 months; however, it is important to note that these differences seem clinically small, with sleep duration changing throughout from about 5 hours to just over 6 hours. Another concern is with regard to the data available in older adults. Although subject ages in this study ranged from 21 to 69 years, the mean age in the eszopiclone group was 44.3 years 11.4, limiting the application of these findings to older adults until further data are available. The association of falls and fractures with eszopiclone has not been studied; however, other hypnotics with similar mechanism of action are associated with falls. Careful counseling of patients and falls risk screening should be implemented whenever using hypnotic agents.
DIABETES
For the treatment of diabetes, several new therapies were submitted for approval in 2004-2005, expanding the potential treatment armamentarium for this illness now affecting more than 20 million persons in the United States.8 An inhaled powdered formulation of insulin (IPI) was approved in January 2006 for use in type 1 and type 2 diabetes mellitus. The availability of this novel dosing formulation of insulin for prandial glucose control is exciting, given the alternative to injections it presents for those with type 2 diabetes who have not achieved good glucose control on oral therapies. In those with type 1 diabetes, IPI may provide a benefit of reduced number of injections throughout the day. In these patients, however, there may be less flexibility in scheduling mealtime coverage than with the currently available short-acting insulin analogs, because the onset of action of IPI is not as rapid. Efficacy and safety of IPI are comparable to other injectable insulins with similar time-action profiles. The pharmacokinetics of IPI blend those qualities seen with regular insulin and the newer, rapid-acting insulin analogs, aspart, lispro, and glulisine. IPI absorption rate or extent in the lung are not specifically known, although 1 mg is therapeutically equivalent to approximately 2-3 units of short-acting insulin with regard to blood glucose-lowering effect. Absorption is altered by cigarette smoking with increased total exposure and maximal concentration.9 The product is not recommended in those who smoke or who quit smoking in the previous 6 months.10 The effects of passive smoke inhalation are not known at this time. After inhalation by healthy volunteers, onset of glucose-lowering activity is more rapid than regular insulin at 10-20 minutes, with peak action earlier than regular at between 30 and 90 minutes.10 Duration of effect, however, is similar to regular insulin (6 hr).11 Elimination is renal, as with other insulins. Dosing of IPI involves use of single-dose blister packs of 1- or 3-mg doses equivalent to 2-3 units or 6-9 units of insulin, respectively, when inhaled. The blister pack is placed into an orally administered pulmonary inhaler. This device disperses as an aerosolized cloud into a holding chamber from which the patient should inhale the dose using a slow, deep breath. It is not clear at this time if multiple doses may be inhaled at once. In clinical trials, the maximum dose used was 92 units, which likely required multiple inhalations. There may also be a difference in the amount of insulin absorbed from the use of three of the 1-mg doses as compared to one blister pack of the 3-mg dose, so substitution is not recommended.
The major adverse effect of IPI is the concern for hypoglycemia, as with other insulin formulations. Insulin antibodies have been demonstrated to occur at a faster rate than for standard injectable insulins; however, this does not appear to be clinically significant. In clinical trials at 4 years, the long-term pulmonary safety of IPI appears good, and this was the subject of extended FDA advisory panel review.12 Safety of IPI in those with pulmonary conditions or using other pulmonary medications has not been established, precluding use in these populations until information is available.
In clinical trials of type 2 diabetes, IPI demonstrated similar efficacy in 149 subjects over 6 months when given prandially with a single bedtime injection of ultralente, as compared to two-dose regimen of regular and NPH insulins. In this study, the mean age of subjects was 58 years in clinical trials with inclusion of subjects 35-80 years old, providing some insight into the use of IPI in older adults.13 In type 1 diabetes, efficacy of IPI given prandially with once-daily ultralente as compared to an NPH am/pm plus pre-meal regular insulin regimen was similar.14 A study recently published by Skyler et al15 further supported comparable efficacy of using IPI in a basal/bolus regimen (pre-meal IPI with twice-daily NPH insulin) in type 1 diabetes, as compared to a standard basal/bolus regimen of injectable insulins (pre-meal regular and twice-daily NPH insulin).
Amylin and Incretin Mimetics
Other exciting new therapies for diabetes include the amylin analogs and incretin mimetics. Agents from each of these categories were approved for use by the FDA this year with the approval of pramlintide, an amylin analog, in March 2005 and exenatide, an incretin (glucagon-like peptide [GLP] mimetic agent), in April 2005. Both pramlintide and exenatide are injectable medications and should not be mixed with insulin. Pramlintide is approved for use in both type 1 and type 2 diabetes, while exenatide is approved for type 2 diabetes.
Pramlintide delays gastric emptying and may regulate glucagon release, resulting in improved postprandial glucose control and centrally-mediated satiety. In clinical trials involving patients with type 1 diabetes, those using the 120-µg dose after 6 months of therapy experienced an absolute A1C reduction of 0.57% from a baseline of 9.1%.16 Patients with type 2 diabetes in another trial experienced a 0.43% absolute reduction in A1C relative to baseline.17 Pramlintide is administered before meals by subcutaneous injection with an initial dose of 15 µg before each meal for those with type 1 DM and an initial dose of 60 µg before each meal for those with type 2 DM. Pramlintide decreases the rate of glucose appearance in the blood, and so may increase the risk of insulin-induced hypoglycemia. Therefore, rapid-acting or short-acting insulin dosages should be reduced by 50% upon initiating pramlintide to prevent hypoglycemia if the patient is using insulin. Blood glucose should be monitored frequently. Once the target dose of pramlintide is reached, insulin doses should be adjusted to optimize therapy. The retail price of pramlintide is approximately $269.91 for a 15-mL vial of 0.6 mg/mL solution (Table). Exenatide is a novel drug modeled after exendin-4, a hormone secreted in the saliva of the Gila monster (Heloderma suspectum), a lizard native to the southwestern United States. In the Gila monster, exendin-4 modulates the ability of the pancreas to turn on and off in relation to the few times the reptile eats throughout the year. A hormone with similar function in humans is GLP-1, which is secreted in the gut and can stimulate glucose-mediated insulin production. Exenatide is similar to human GLP-1. There are four proposed mechanisms in which exenatide exerts its glucose-lowering effects: improves glucose-dependent insulin secretion, moderates glucagon secretion, slows gastric emptying, and reduces food intake.

When exenatide is used in combination with metformin, mean absolute A1C reductions at week 30 were 0.4% (≤ 0.05 vs placebo) for the group receiving 5 µg twice daily and 0.8% (≤ 0.001 vs placebo) for the group receiving10 µg twice daily.18 Mean change in body weight at week 30 was -1.6 kg (≤ 0.05 vs placebo) and -2.8 kg (≤ 0.001 vs placebo) in the 5-µg and 10-µg groups, respectively. When administered with a sulfonylurea, absolute mean reduction of A1C at week 30 was 0.5% (≤ 0.05 vs placebo) and 0.9% (≤ 0.001 vs placebo) for 5 µg twice daily and 10 µg twice daily, respectively.19 Mean body weight reductions of -0.9 kg in the 5-µg group (NS) and -1.6 kg in the 10-µg group (≤ 0.05 vs placebo) were realized. When exenatide was used in combination with both metformin and a sulfonylurea, mean absolute A1C reductions at week 30 were 0.6% and 0.8% for the 5- and 10-µg groups, respectively (both ≤ 0.001 vs placebo).20 Both the 5- and 10-µg groups experienced a -1.6 kg mean body weight loss (both ≤ 0.05 vs placebo). Exenatide is administered twice daily as a 5-µg subcutaneous injection within 60 minutes before the morning and evening meals. The retail price of exenatide, available in a prefilled pen-dosing cartridge, is approximately $196.98 for a 2.4 mL-cartridge of 250-µg/mL solution.
Pramlintide and exenatide share a similar side-effect profile of hypoglycemia and gastrointestinal disturbances (eg, nausea, vomiting, diarrhea, dyspepsia). In clinical trials with older patients using pramlintide (n = 539), no consistent age-related differences in activity were noted. Exenatide has been studied in close to 300 patients 65 years of age or older, and in only 16 patients 75 years of age and older. Safety and effectiveness in these older patients did not differ from younger patients. However, in older patients, extra careful attention to blood glucose levels through monitoring should be given during therapy initiation and dosage changes with pramlintide or exenatide.
Insulin Detemir
Currently available basal insulins include NPH, insulin glargine, and ultralente. Most patients require twice-daily dosing of NPH and once-daily dosing of ultralente and glargine to meet their basal insulin requirements. The new recombinant basal insulin detemir was approved by the FDA on June 16, 2005; however, detemir has been available in some European countries since 2004.
Insulin detemir was developed by covalently bonding a 14-carbon fatty acid (mysteric acid) to the amino acid lysine at postion B29 while omitting threonine, the amino acid at position B30 on the human insulin molecule. These modifications result in an insulin that has a high albumin-binding affinity, which results in prolonging the release from the subcutaneous space and prolonging the action of detemir in the plasma. Insulin detemir is reversibly bound to albumin (97-99%). Although detemir is highly protein-bound, it has not been shown to interact with other highly protein drugs such as warfarin, valproate, or ibuprofen.21 This lack of interaction most likely occurs because binding of insulin detemir to albumin is independent of ligand binding at the IIA domain of albumin. Therefore, reduced albumin levels do not increase the risk of interaction; however, this has not been specifically studied clinically. The peak concentration of detemir is attained in 6-8 hours post-injection, and most patients will require twice-daily dosing. The initial starting dose is in the range of 0.2-0.5 U/kg/day with titrations based on blood glucose levels. As with all insulin, dosing will need to be individualized.
Potential advantages of detemir over NPH insulin include less within-subject variability, similar A1C-reducing capability, less weight gain, and less hypoglycemia. In a study by Vague et al22 involving 448 patients with type 1 diabetes, less within-subject day-to-day variation in fasting self-monitoring of blood glucose was noted than with NPH insulin (P < 0.001). In another study, hemoglobin A1C reduction over 16 weeks was studied in 408 patients with type 1 diabetes. Subjects were given premeal aspart and either insulin detemir as a twice-daily injection with breakfast and at bedtime, or an every-12-hour injection of detemir or NPH given prior to breakfast and at bedtime. In this study, there was no statistically significant difference in A1C between the groups at study end (ANOVA, P = 0.082).23 Multiple studies have examined detemir’s effects on weight gain. The aforementioned studies by Vague et al22 and Home et al23 both demonstrated a statistically significant effect on weight gain as compared to NPH. In the study by Vague et al,22 patients taking detemir experienced a 0.2-kg weight loss as compared to a 0.7-kg weight gain with NPH (P = 0.001) over the 6-month study.
Hypoglycemia is the most frequently encountered side effect in insulin-requiring patients. In multiple studies, the incidence of hypoglycemia has been shown to be lower in patients using detemir in comparison to NPH. In the study by Vague et al,22 the risk of hypoglycemia was 22% lower with detemir than with NPH (hazard ratio, 0.78; 95% CI: 0.62- 0.97; P < 0.001), and the risk of nocturnal hypoglycemia was 34% lower in the detemir group as compared to the NPH cohort (hazard ratio, 0.66; 95% CI: 0.50-0.87; P < 0.005).
In summary, insulin detemir may be a viable alternative for insulin-requiring patients who are not meeting A1C goals with NPH insulin and/or are experiencing untoward side effects such as weight gain or hypoglycemia. While no studies have specifically studied detemir in older populations, the same precautions with older insulin-requiring patients with diabetes are advised (eg, frequent self-monitoring of blood glucose, education on hypoglycemic signs and symptoms). As with all new therapies, the best clinical use of exenatide and pramlintide needs to be further elucidated, and specific study in geriatric patients with diabetes would assist in determining the risk/benefit of these antidiabetic therapies in the elderly.
CONCLUSION
There are many new and exciting therapies available for the treatment of OAB, insomnia, and diabetes. As with most new drugs, caution is advised when using these newer agents in older adults, and it is reasonable to delay the use of newer agents until some time has passed and experience has been gained using these drugs on younger, and possibly healthier, adults. In addition, when using newer agents in older adults, greater vigilance for side effects and screening for drug interactions is warranted, given the often limited use in this population at the time of marketing.
Acknowledgments
The authors would like to thank Joyce Harbison, assistant with the Washington State University College of Pharmacy, for her editorial assistance with this article.
Presentation of information in this article was sponsored in part by AARP at the 2005 AGS Annual Scientific Meeting. The authors report no relevant financial relationships.
