


A Case Study of IdiopathicThrombocytopenic Purpura
Case Presentation
A 72-year-old long-term functional nursing home resident with a history of small cell lung cancer diagnosed 5 months previously was transferred to the emergency room with a chief complaint of 1 week of severe generalized weakness and moderate shortness of breath, as well as heavy epistaxis for 24 hours. The patient had received eight administrations of cisplatin (etoposide) as part of his regimen for cancer treatment, the most recent one 3 weeks ago. The patient also related he’d bitten the inside of his mouth about 1 week ago, and he still had mild, occasional bleeding from the site, despite being extremely careful when he chewed food.
On review of systems, the patient related he had been suffering from chronic dizziness for several months since he’d started his chemotherapy, along with blurred vision and a nonproductive cough. He still had a good appetite but no bowel movement in the last 2 days. He denied any other complaints.
Past medical history included two cerebrovascular accidents, colon polyps, peripheral vascular disease, and past exposure to asbestos when he worked as a pipe fitter many years ago. He had been a heavy smoker, averaging 1 pack per day for 50 years until quitting 1 year ago. Family history was noncontributory. His allergies included only penicillin.
Investigation
On physical examination, the patient was an elderly man in no apparent distress but obviously weak. His height and weight were appropriate for his age. His blood pressure was 86/55 mm Hg, and he was afebrile. Lungs were clear, and there was no hepato- or splenomegaly. Neurological evaluation was normal. His skin was dry and turgor was reduced. Orthostatics were positive. There was a round, inflamed lesion of dried blood located on the right buccal mucosa. His skin revealed diffuse petechia on the bilateral lower extremities up to the knees, as well as across the lower abdomen.
Laboratory work-up showed a complete metabolic panel that was within normal limits. The white cell count was 7200/μL, hemoglobin and hematocit were 10.2 g/dL and 31.2 g/dL, respectively, and platelet count of only 1/μL. The differential showed a granulocyte count of 81% and lymphocyte count of only 9%. A chest x-ray was essentially unchanged from 6 months earlier, indicating the presence of a soft tissue mass in the left lower extremity with some pleural thickening, along with fibrosis in the right upper lobe.
The patient was admitted to the hospital with a diagnosis of small cell lung cancer, hypotension, and severe thrombocytopenia. Orders were written for the patient to begin receiving a transfusion of platelets, as well as being started on prednisone empirically, intravenous fluids, and multivitamins. A work-up for hemolysis and disseminated intravascular coagulation (DIC) was begun.
 A peripheral blood smear did not reveal any abnormalities in the structure of the red blood cells: there were no multinucleated forms, teardrop formations, or schistocytes. Few platelets were seen on the slide (Figure 1), and there was no hemolysis. The D-dimer, fibrinogen, and fibrin degradation products were all negative. Sedimentation rate, C-reactive protein, and coagulation parameters were normal. Lactate dehydrogenase and uric acid were elevated at 275 U/L and 8.2 mg/dL, respectively. Thyroid and renal function were normal. Workup for autoantibodies (ie, antinuclear antibody, rheumatoid factor, and antimicrobial antibody) was conducted; however, none of these were detected.Within 48 hours the patient had received a total of 20 units of platelets.
A peripheral blood smear did not reveal any abnormalities in the structure of the red blood cells: there were no multinucleated forms, teardrop formations, or schistocytes. Few platelets were seen on the slide (Figure 1), and there was no hemolysis. The D-dimer, fibrinogen, and fibrin degradation products were all negative. Sedimentation rate, C-reactive protein, and coagulation parameters were normal. Lactate dehydrogenase and uric acid were elevated at 275 U/L and 8.2 mg/dL, respectively. Thyroid and renal function were normal. Workup for autoantibodies (ie, antinuclear antibody, rheumatoid factor, and antimicrobial antibody) was conducted; however, none of these were detected.Within 48 hours the patient had received a total of 20 units of platelets.
Discussion
Thrombocytopenia is defined as a quantitative dysfunction of the platelets, either by an absence or a decrease in their number and thus an inability to operate normally (ie, participate in clotting). This typically causes a prolongation in bleeding time, but coagulation parameters are within normal limits. The bone marrow reveals either a reduction in the percentage of megakaryocytes (if platelet production is the problem) or increased megakaryocytes (if there is increased destruction of platelets). Low platelet numbers are due to reduced production, increased destruction, unreplaced loss, or dilution.1
A platelet count of 100,000/μL is considered thrombocytopenia, but spontaneous bleeding doesn’t typically occur until the count is below 20,000, with severe hemorrhage risk increasing when the count goes below 10,000. In this case, steroids, plasmapheresis, or transfusion may be needed. A decrease in platelet count stimulates an increase in thrombopoietin, which produces an increase in platelet production.
 There are three main etiologies of thrombocytopenia: (1) production, (2) destruction, and (3) dilution (Table I). Production deficits causing thrombocytopenia are due to: viral infections (eg, cytomegalovirus, rubella, Epstein-Barr virus, human immunodeficiency virus, hepatitis, parvovirus B19); immunizations; medications (note that in most cases, a cause-and-effect relationship has not been demonstrated; common offenders include histamine [H2]-blockers, antiepileptics, and antibiotics). Destruction includes the effects due to DIC and idiopathic thrombocytopenic purpura (ITP).
There are three main etiologies of thrombocytopenia: (1) production, (2) destruction, and (3) dilution (Table I). Production deficits causing thrombocytopenia are due to: viral infections (eg, cytomegalovirus, rubella, Epstein-Barr virus, human immunodeficiency virus, hepatitis, parvovirus B19); immunizations; medications (note that in most cases, a cause-and-effect relationship has not been demonstrated; common offenders include histamine [H2]-blockers, antiepileptics, and antibiotics). Destruction includes the effects due to DIC and idiopathic thrombocytopenic purpura (ITP).
The work-up for thrombocytopenia includes assessing whether the cause is drug-induced, or due to a certain disease process (ie, thrombotic thrombocytopenic purpura [TTP], hemolytic uremic syndrome, or ITP).
The mechanism of drug-induced thrombocytopenia involves either toxicity to the bone marrow or platelet destruction in the peripheral blood. The most common offenders are listed in Table II. Over-the-counter vitamin preparations and health supplements may also elicit the condition. In such situations, treatment involves the immediate cessation of the causal agent. Platelet counts should increase to normal or near normal within 7-10 days, except with gold where bone marrow damage requires months to recover. Laboratory tests can identify the causative agent in 10% of patients with drug-induced thrombocytopenia.2
TTP typically occurs in young and middle-aged women. Its cause is unknown, but symptoms include the typical pentad of renal failure, fever, microangiopathic hemolytic anemia, thrombocytopenia, and neurologic symptoms. Widespread hyaline microthrombi occur, and schistocytes may appear on the peripheral smear. Thrombocytopenia can also be due to the following: AIDS, systemic lupus erythematosis (SLE), infectious mononucleosis, toxoplasmosis, cytomegalovirus, and lymphoproliferative disorders.
 Signs and symptoms of thrombocytopenia include petechia, mucosal surface oozing, and intracranial, gastrointestinal and urinary tract bleeding. With thrombocytopenia, bleeding time is prolonged in TTP and DIC, but not in ITP. Prothrombin time (PT), partial thromboplastin time (PTT), thrombin, and fibrinogen assays are also normal in ITP.
Signs and symptoms of thrombocytopenia include petechia, mucosal surface oozing, and intracranial, gastrointestinal and urinary tract bleeding. With thrombocytopenia, bleeding time is prolonged in TTP and DIC, but not in ITP. Prothrombin time (PT), partial thromboplastin time (PTT), thrombin, and fibrinogen assays are also normal in ITP.
ITP has also been called immune or autoimmune thrombocytopenic purpura. It is a hematologic disorder occuring in children and older adults. In children, it is often acute and self-limited due to a viral infection (ie, upper respiratory) or immunization. Sixty percent of children recover spontaneously within 4-6 weeks, and over 90% within 3-6 months.2,3 In adults, it takes on a chronic, indolent form that may last for years. Less than 10% of cases in adults resolve spontaneously.4 It is not caused by drugs or any other identifiable disorders (Table II). ITP occurs most often in adult females between 20 and 40 years old, and mothers have been known to pass on the autoantibodies to their children via placental crossing. The female-to-male ratio is 3:1. There is no apparent racial or environmental predilection.
The mechanism of pathology of ITP involves the generation of autoimmune antiplatelet antibodies (typically immunoglobulin G [IgG], produced from B cells usually within the spleen). The Fc portion of these antibodies coat platelets and cause complement activation, thus damaging the platelet membranes. These platelets are selectively and prematurely removed or cleared in the reticuloendothelial system (RES) via phagocytosis by splenic macrophages. Platelet survival is reduced from the normal 7-10 days to several days or even hours. Of note is that the spleen is capable of holding one-third of the total circulating pool of platelets in the body.4 In this condition, the bone marrow does increase platelet production but is unable to adequately compensate for the loss in numbers.
The majority of patients with ITP have been found to have autoimmune immunoglobulins that specifically attack antigens on the platelet membrane glycoproteins (Gp) Ib or IIb/IIIa complexes.5 Some antibodies have been found to bind megakaryocytes, and thereby inhibit platelet production. This condition is usually an isolated derangement separate from other autoimmune disorders, such as SLE or thyroid disease. On history, rule out taking sulfonamides, aspirin, and anticoagulants.
Symptoms of ITP include ecchymoses, petechiae, purpura, epistaxis, and bleeding and hemorrhage from mucosal and serosal membranes and various areas of the body (gingival, rectal, vaginal). There may be a history of easy bruising or menometrorrhagia. There is usually no splenomegaly.
In working up thrombocytopenia, rule out alcohol use; recent blood transfusions within 1-2 weeks, which can illicit a purpuritic reaction; viral illness; liver disease; HIV risk factors (ie, intravenous drug abuse, sexually transmitted diseases); and exposure to certain toxins (eg, ionizing radiation, benzene, insecticides, nitrogen mustard). Investigation of inherited disorders such as Bernard-Soulier Disease, Wiskott-Aldrich syndrome, and Von Willebrand disease may be indicated in younger patients. On physical examination, assess for skeletal deformities such as absence of the radius, which indicate congenital thrombocytopenia. Also determine whether hepato- and/or splenomegaly is present, as well as lymphadenopathy (may suggest cancer, infection, or lymphoproliferative disease). Splenomegaly suggests ITP is not present.
Some studies have shown a link between Helicobacter pylori infection and the occurrence of ITP.6 More specifically, eradication therapy to treat H. pylori appears to decrease platelet activation; however, platelet counts remain unchanged.
Chronic ITP may by related to tuberculosis, sarcoidosis, SLE, lymphoma, chronic lymphocytic leukemia, and Evans syndrome.4 The lifespan of some patients with chronic ITP may be up to two to three decades.
The occurrence of ITP may have atypical etiologies in some patients, such as with influenza vaccine7, interferon (IFN), or pegylated interferon (PEG-IFN) alfa 2a therapy used in the treatment of chronic hepatitis.8-10 In such cases, the patient may have the condition due to an unknown cause during the initial evaluation, but after determination of the etiology, a designation of immune or autoimmune thrombocytopenia is more accurate and appropriate.
Diagnosis
 A diagnosis of ITP is made after other disorders and etiologies have been ruled out. The work-up involves testing a peripheral blood smear and possibly performing a bone marrow aspirate and biopsy (Figures 1 and 2). The biopsy should reveal a low number of platelets without clumping (pseudothrombocytopenia), normal or elevated megakaryocytes, and normal leukocyte and erythrocyte counts and morphology. This is done if prior studies are inconclusive or abnormalities are present on the blood smear, or in elderly patients who are positive for ITP and show normal or increased numbers of megakaryocytes. A bone marrow biopsy is also performed if considering splenectomy in order to verify the diagnosis, as well as to exclude a myelodysplastic syndrome, which often occur in the elderly.11 A test to determine the presence of platelet-associated IgG is also available.
A diagnosis of ITP is made after other disorders and etiologies have been ruled out. The work-up involves testing a peripheral blood smear and possibly performing a bone marrow aspirate and biopsy (Figures 1 and 2). The biopsy should reveal a low number of platelets without clumping (pseudothrombocytopenia), normal or elevated megakaryocytes, and normal leukocyte and erythrocyte counts and morphology. This is done if prior studies are inconclusive or abnormalities are present on the blood smear, or in elderly patients who are positive for ITP and show normal or increased numbers of megakaryocytes. A bone marrow biopsy is also performed if considering splenectomy in order to verify the diagnosis, as well as to exclude a myelodysplastic syndrome, which often occur in the elderly.11 A test to determine the presence of platelet-associated IgG is also available.
The acute onset of petechiae, ecchymoses, and bleeding may suggest idiopathic thrombocytopenia (often associated with a recent viral infection, large platelets in blood smears, and no evidence of anemia).
Treatment
Unlike in children, the majority of whom recover spontaneously, ITP usually persists in adults and will eventually require some type of medical intervention. The method of treatment depends on the degree of symptomatology and thrombocytopenia.
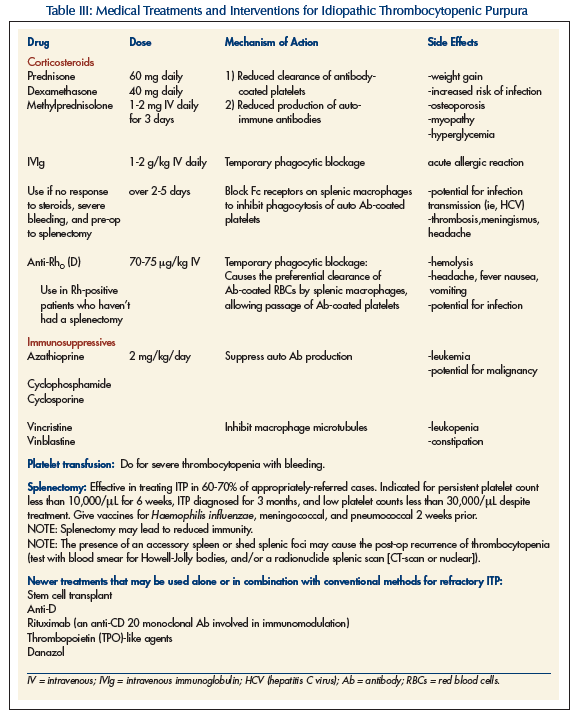
Treatment is with steroids, intravenous immunoglobulin (IVIg), Anti-Rho (D) – known as RHO immune globulin, danazol, and immunosuppressives (eg, vincristine, azathioprine, cyclophosphamide). Splenectomy may be needed if medical treatment is ineffective. It may also be required emergently if internal bleeding is present (especially intracranial) and with platelets less than 30,000\μL. Transfusion of platelets would generally be useless, given that the autoantibodies to the platelets are still present within the bloodstream. Such a transfusion is only used in the event of intracerebral bleeding.
Patients with bleeding or platelet counts less than 20,000 should be hospitalized. Hospitalization is not needed if the patient is asymptomatic and the platelet count is greater than 20,000, or if the patient has only minor bleeding.
Consult the Hematology/Oncology service if there is severe thrombocytopenia or abnormal changes in leukocyte or erythrocyte morphology or counts. In addition, do a consult if anemia is present without iron deficiency or blood loss, or if myeloid precursors are found on the peripheral smear.
Corticosteroids are regarded as the first line of treatment for ITP in adults, utilizing their immunosuppressive role in an autoimmune condition. Various types can be used, from prednisone to dexamethasone (Table III). Steroids along with IVIg are considered conventional treatments.
Anti-Rho (D) immunoglobulin was first licensed in Canada in 1980 for the prophylaxis of hemolytic disease in neonates due to Rh isoimmunization.8,9,10 It is a gamma immunoglobulin G that recognizes and binds specifically to the erythrocyte D antigen on red blood cells (RBCs). It works only in nonsplenectomized Rh-positive patients and is administered intravenously in doses of 20-100 μg/kg. Its mechanism of action is not completely understood, but is thought to involve immunoglobulin attachment to erythrocytes which eventually travel into the RES. These coated RBCs are preferentially cleared by the spleen, causing some hemolysis but saturating the FcR portions of splenic macrophages that are responsible for latching onto and removing antibody-coated platelets. If enough of these sites are occupied by anti-Rho (D)-coated RBCs, antibody-coated platelets can pass through the RES and survive. Common drug reactions to anti-Rho (D) include mild to moderate headache, fever, chills, dizziness, nausea and vomiting, myalgias, hemolysis, and a positive Coombs’ reaction.
Some immunosuppressive drugs used in cancer treatment are administered in certain cases of ITP refractory to the conventional interventions. Danazol, an anabolic steroid, has also been found to be a useful treatment in select cases of refractory ITP. New treatments for ITP include anti-D, thrombopoietin-like medications, and rituximab (Table III).12 Stem cell transplantation is also being investigated.
Patients who don’t respond to initial treatment of ITP—or those who relapse—should be tested for H. pylori. This is done with a C13 urea breath test or serum test.
Consider splenectomy if the patient doesn’t respond to steroids, or if relapse occurs. Such conditions involve persistently low platelet counts of less than 10,000 for 4-6 weeks, and patients diagnosed with ITP for at least 3 months with platelet counts less than 30,000 and no response to treatment. Over two-thirds of patients who undergo splenectomy experience complete remission of the disease with a return to normal platelet counts. Patients with this condition should avoid contact sports, as well as alcohol use, aspirin, nonsteroidal anti-inflammatory drugs, and anticoagulants.
In deciding upon a treatment modality, the medical provider must keep in mind the potential toxicity that may result. In addition, realize that there is currently no medical consensus or formalized algorithm delineating systematic treatment methods for ITP. However, the majority of appropriately treated patients do recover.
Outcome of the Case Patient
Additional lab work did eventually confirm the presence of antiplatelet antibodies. This finding, along with the clinical presentation, were consistent with a diagnosis of ITP. A bone marrow biopsy was deemed unnecessary. The patient was started on oral prednisone 1mg/kg and, after discharge from the hospital, was followed at regular intervals by the outpatient Hematology service. His platelet count progressively increased until it had reached baseline a month later, at which time the steroid was tapered.
The symptoms with which this patient presented were subtle, and in the elderly population could have represented a spectrum of conditions, from a state of dehydration and malnutrition to iron deficiency to a complication of his previously diagnosed cancerous process. It was also important to realize that this patient was a functional and ambulatory nursing home resident, and that his symptoms could have possibly been due to not only cancer but also a reaction to the treatment he had received for it. ITP in this patient, being male and presenting at this age, is rare. The finding of a severely low platelet count on lab studies was crucial to making a diagnosis and implementing appropriate medical treatment in a timely manner.
The author reports no relevant financial relationships.
