Initial Results of Inflammatory Response, Matrix Remodeling, and Reactive Oxygen Species Following PCI in Acute Ischemic Myocardial Injury in Man
ABSTRACT: Background. Neutrophils and reactive oxygen species (ROS) are suggested to be involved in irreversible myocardial reperfusion injury and stunning. We investigated the relations between circulating biochemical markers and myocardium at risk (MaR), myocardial infarct (MI) size, salvage, and recovery of function in man. Methods and Results. In patients undergoing PCI serial blood samples were acquired for markers of inflammatory response (myeloperoxidase [MPO], neutrophil-gelatinase-associated lipocalin [NGAL], interleukins 6 and 8 [IL-6/8], tumor necrosis factor-α [TNF-α], high-sensitive C-reactive protein [hsCRP]), matrix remodeling (matrixmetalloproteinase-9 [MMP-9]) and ROS (malondialdehyde [MDA], isoprostane [IsoP]). Samples were obtained before PCI and 1.5, 3, and 24 hours after reperfusion. Myocardial perfusion SPECT (MPS) was used to assess MaR. Late gadolinum-enhanced cardiac magnetic resonance imaging was performed for regional function in the acute setting, at 1 week and 6 months, and at 1 week also for MI size. Sixteen patients (15 men; 42–78 years) were enrolled, 12 of whom underwent MPS. Peak and cumulative NGAL and cumulative MMP-9 showed inverse correlations to MaR. No correlation was found for MI size. Peak MPO correlated inversely to salvage and to recovery of regional function in the infarcted segments at 1 week and 6 months. Conclusions. This is the first study in man to show inverse relations between circulating NGAL and MMP-9 and MaR. The current results do not support that ROS has a role in stunning in man. MI size showed no significant correlation to any parameter, challenging inflammatory treatment in reperfusion.
J INVASIVE CARDIOL 2011;23:371-376
Key words: biomarkers; salvage, myocardial; stunning, myocardial; myocardium at risk; infarct size, myocardial
__________________________________
Even though early reperfusion after coronary artery occlusion in humans reduces myocardial infarct (MI) size1 and mortality,2 reperfusion may also induce damage to the myocardium in itself.3,4 Both stunned myocardium and irreversible reperfusion injury have been proposed to be caused by cellular dysfunction, either via formation of reactive oxygen species induced by proteolytic enzymes,5,6 or by neutrophils and their toxic degradation products accumulated in the ischemic as well as reperfused myocardium.7 In animals, experiments have shown a decreased MI size by using anti-inflammatory treatments such as corticosteroids, complement-depletion, nonsteroidal anti-inflammatory drugs, or inhibiting arachidonic metabolism pathways.8–10 The transfer of animal findings to man has, however, been unsuccessful or even increased MI size and ventricular arrhythmias.11–14 The reason for this is unknown, and it is thus of significant interest to study inflammatory markers in man following reperfusion by PCI. The inflammatory response is activated early, before restoring flow by percutaneous coronary intervention (PCI).15–17 Therefore, myocardium at risk is warranted as a measure of the myocardium subject to ischemia in acute coronary syndrome.
Reactive oxygen species and inflammatory response are feasible to indirectly measure by malondialdehyde and isoprostanes; and myeloperoxidase, neutrophil gelatinase-associated lipocalin,8 interleukin 6 and 8,15 tumor necrosis factor α,19 high-sensitive C-reactive protein,20 and matrix metalloproteinase,21 respectively.
The aim of this study was therefore to investigate how peak and cumulative levels of circulating markers of inflammatory response, matrix remodeling and reactive oxygen species, relate to myocardium at risk by myocardial perfusion single photon emission computed tomography, final MI size by cardiac magnetic resonance (CMR) imaging, salvage of myocardium at risk, and to recovery of regional function in patients with acute coronary occlusion, treated by PCI.
Materials and Methods
Study population. The study was approved by the local ethics committee and conforms with institutional guidelines and those of the American Physiological Society and the Helsinki Declaration. Written consent was obtained from all patients. To be included, patients should have no history of previous MI, a single occluded (TIMI 0) artery followed by successful revascularization by primary PCI (pPCI) (TIMI 3) and absence of collateral flow by angiography. Furthermore, patients should have no release of markers of MI before pPCI. Patients that were transferred to acute or subacute surgery were excluded. Some of the patients included in this study (n = 8) have been part of a previous study.1
All patients were pretreated with 300 mg acetylsalicylic acid (ASA) and 300 mg clopidogrel prior to the procedure. At least one bare-metallic stent was used in each patient. All patients also received the integrin glycoprotein IIb/IIIa receptor inhibitor (GP IIb/IIIa) abciximab, administered as a bolus of 0.25 mg/kg of body weight during the pPCI, followed by a continuous 10 μg/minute infusion for 12 hours post intervention. Unfractionated heparin was administered during pPCI to achieve an activated clotting time (ACT) of 250 s. The ACT was measured using a Hemochron system. Diagnostic coronary angiography for inclusion in the study was performed immediately before pPCI. After identification of the culprit lesion, intervention was initiated with a 6 Fr catheter via the femoral artery approach. Angiography after intervention confirmed TIMI 3.
Drugs used during intervention were intracoronary nitroglycerin, intravenous beta-blocker agents, intravenous morphine-hydrochloride, dixyrazin, diazepam, and intravenous Ringer-Acetate or glucose.
Study protocol. Serial blood samples for analysis of biochemical markers of inflammatory response were obtained prior to opening of the occluded infarct-related vessel, and at 1.5, 3, and 24 hours post reperfusion. For determination of myocardium at risk, 99 mTc-tetrofosmin was injected approximately 5 minutes prior to opening of the occluded coronary artery by pPCI, and myocardial perfusion SPECT was performed within 3 hours thereafter. All patients were transferred post pPCI to the coronary care unit for conventional therapy, and had an uncomplicated clinical course. The CMR was performed the same day for baseline regional function, at 1 week for initial functional recovery and MI sizing, and at 6 months for assessment of functional recovery. Measurement of MI size was performed at 1 week after MI in order to avoid overestimation of MI size.22
Regional function was calculated from cine MR images as wall thickness in systole versus diastole in 72 segments, defined as the heart divided into 6 short-axis slices, each with 12 circumferential sectors. Segments were defined as infarcted, adjacent or remote to infarction and analyzed for recovery in function. Salvage was calculated as 100 x ([myocardium at risk]-MI)/[myocardium at risk].
Biochemical markers. Samples for determination of levels of circulating biochemical markers consisted of venous blood, except for the baseline sample which was arterial and obtained via the arterial sheath before reperfusion. All samples were obtained in tubes with ethylenediaminetetraacetic acid (EDTA), either with citrate or without additives (Becton Dickinson). The EDTA tubes were used for P-myeloperoxidase, P-interleukin 6 and P-interleukin 8, P-tumor necrosis factor-α, P-malondialdehyde, and P-isoprostane. Tubes without additives were used for S-neutrophil-gelatinase-associated lipocalin, S-matrix-metalloproteinase-9, and S-high-sensitive C-reactive protein. They were all centrifuged at 2000 G at 4 ºC, cooled, and stored at -70 ºC before the respective assay. Analyses were performed up to 4 years after acquisition, except for malondialdehyde, which was analyzed within a few weeks from sampling. Levels of circulating P-myeloperoxidase, S-neutrophil-gelatinase-associated lipocalin, S-matrix-metalloproteinase-9, P-interleukin 6 and P-interleukin 8, P-tumor necrosis factor-α, S-high-sensitive C-reactive protein, P-malondialdehyde, and P-isoprostane were analysed as previously described.15 In short, commercially available methods using either high-performance liquid chromatography (HPLC), enzyme-linked immunosorbent assay (ELISA) or particle-enhanced immunoturbidimetric assays (PEITA) were utilized.
Myocardial perfusion SPECT. Patients were injected with 500-700 MBq 99mTc tetrofosmin (Amersham Health), depending on bodyweight. The ECG-gated myocardial perfusion SPECT was performed according to the standard clinical protocol at rest, using a dual head-camera (ADAC Vertex). The patients were placed in supine position and imaged in steps of 5.6 degrees using a 64 × 64 matrix with a pixel size of 5.02 mm. Image acquisition time was approximately 15 minutes. Short- and long-axis images gated to the ECG were reconstructed using a commercial application (AutoSpect+InStill™ 6.0, ADAC). Analysis of the perfusion defect for myocardium at risk was performed using an FDA-approved, freely available in-house developed segmentation software (Segment v1.839; https://segment.heiberg.se).23 The myocardium at risk was determined in percent of the left ventricle by an in-house developed automated algorithm that considers myocardium with < 55% of normal counts as being subject to ischemia, taking into account the different coronary perfusion territories.24 In short, the automatic segmentation finds the centerline through the left ventricular wall and identifies the endo- and epicardium based on an individually estimated wall thickness and signal intensity values within the image. Manual adjustment of the automatic delineation was sometimes required in the left ventricular outflow region.
CMR. Patients were imaged in the supine position using either a 1.5 T system with a CP body array coil (Magnetom Vision, Siemens), or a 1.5 T system with a cardiac synergy coil (Philips Intera CV, Philips). Acquisition was performed during breath-hold, using ECG-triggered sequences. For determination of the left ventricular regional function, cine short-axis images covering the left ventricle was acquired, using either a gradient-recalled echo (GRE) turbo fast low angle shot (FLASH) sequence (1.6 × 1.6 × 8 mm, slice gap 2 mm, TR/TE 4.8/100 ms) or a balanced single-shot free precession (SSFP) sequence (1.6 × 1.6 × 8 mm, 1.6/3.1 ms). Regional function was calculated from short-axis cine images as wall thickness in systole versus diastole in 72 segments.
For determination of MI size, short-axis late gadolinium enhancement (LGE) inversion-recovery MR images (Siemens: 1.4 × 1.4 × 10 mm, 8/4 ms, flip angle 25°, every other heartbeat; Philips: 1.5 × 1.5 × 8 mm, 3.9/1.2 ms, flip angle 15°, every heartbeat) were acquired 20–30 minutes after administration of 0.2 mmol/kg of body weight of an extracellular gadolinium-based contrast agent (gadoteric acid, Gd-DOTA, Guerbet, Gothia Medical AB). The inversion time of typically 200–350 ms was manually adjusted to null the signal from viable myocardium. The MI quantification was performed on short-axis LGE images using the same software as for the myocardial perfusion SPECT images, and expressed as percentage of the left ventricle. A previously described automatic algorithm for MI border detection was used.25 The MI borders were visually controlled and adjusted if the computer algorithm was obviously wrong.
Statistical analyses. MatLab 7.6.0 R2008a (Mathworks Inc.) was used for calculating the Pearson correlation coefficients and linear regressions for biochemical marker data as functions of myocardium at risk, MI size, salvage, and recovery of regional function. A binomial test was applied to test for global differences regarding correlations between the studied biochemical markers versus myocardium at risk and MI size, respectively. Input data was the absolute r-value for each biochemical marker. A p-value < 0.05 was considered statistically significant. Considering the risk of mass significance, 1 out of 18 comparisons between the markers studied and myocardium at risk might falsely be found statistically significant at the applied level of significance.
Results
 Sixteen patients (15 men; 42–78 years), of whom 12 (all men) also underwent myocardial perfusion SPECT in the acute setting, were prospectively included. Patient characteristics are presented in Table 1. In one patient, analysis of neutrophil-gelatinase-associated lipocalin, matrix metalloproteinase-9, and high-sensitive C-reactive protein could not be performed. The malondialdehyde and isoprostane were not assessed in 4 and 1 patients, respectively, due to technical reasons.
Sixteen patients (15 men; 42–78 years), of whom 12 (all men) also underwent myocardial perfusion SPECT in the acute setting, were prospectively included. Patient characteristics are presented in Table 1. In one patient, analysis of neutrophil-gelatinase-associated lipocalin, matrix metalloproteinase-9, and high-sensitive C-reactive protein could not be performed. The malondialdehyde and isoprostane were not assessed in 4 and 1 patients, respectively, due to technical reasons.
 Baseline. For baseline values of biochemical markers, as acquired via the arterial sheath before restoring flow in the occluded vessel, a significant correlation could not be shown for either myocardium at risk (|r| < 0.6, all p = ns) or MI size (|r| < 0.4, all p = ns).
Baseline. For baseline values of biochemical markers, as acquired via the arterial sheath before restoring flow in the occluded vessel, a significant correlation could not be shown for either myocardium at risk (|r| < 0.6, all p = ns) or MI size (|r| < 0.4, all p = ns).
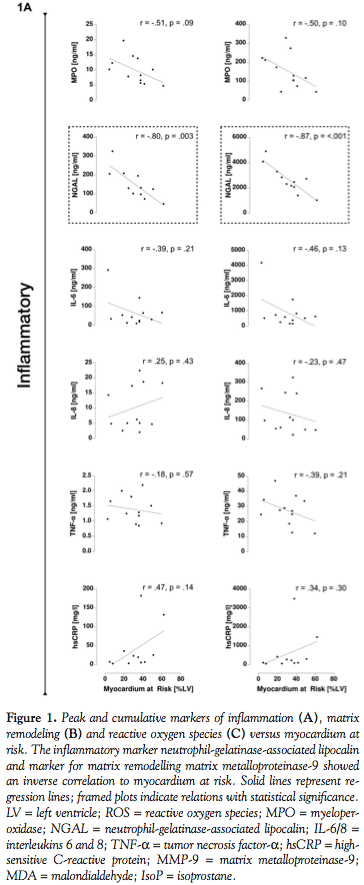
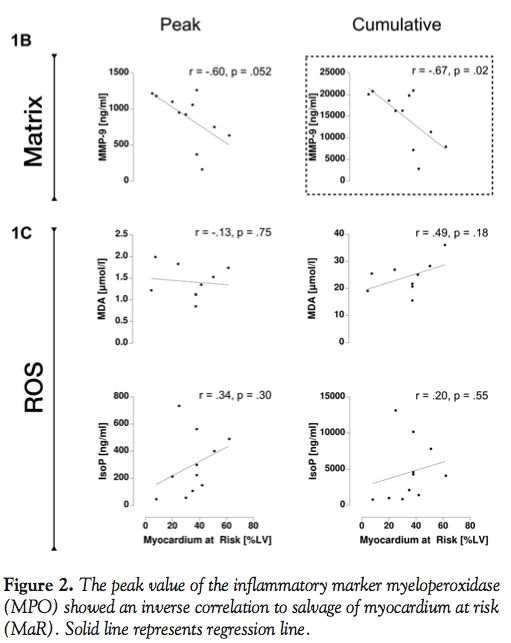
Myocardium at risk, MI size, and salvage. Scatterplots and linear regressions are shown in Figure 1. Statistically significant relations were found between myocardium at risk and neutrophil-gelatinase-associated lipocalin (peak: r = -0.80 and p = 0.003; cumulative: r = -0.87 and p < 0.001) and cumulative matrix metalloproteinase-9 (r = -0.67 and p = 0.02). For peak matrix metalloproteinase-9 and myocardium at risk, a value close to the limit of significance was found (r = -0.60 and p = 0.052).
 Correlations between peak or cumulative markers and MI size by CMR were found to be low and not significant, |r| < 0.50 for all markers (all p = ns). Further, all inflammatory markers studied, with the exception of high-sensitive C-reactive protein, showed an inverse relationship to MI size.
Correlations between peak or cumulative markers and MI size by CMR were found to be low and not significant, |r| < 0.50 for all markers (all p = ns). Further, all inflammatory markers studied, with the exception of high-sensitive C-reactive protein, showed an inverse relationship to MI size.
Salvage of myocardium at risk showed a slightly higher and statistically significant correlation only with peak myeloperoxidase (r = -0.75 and p = 0.003), which was found to show lower values with increased salvage (Figure 2).
The correlation coefficients were generally higher for biochemical markers and myocardium at risk, compared to biochemical markers and MI size (p = 0.007).
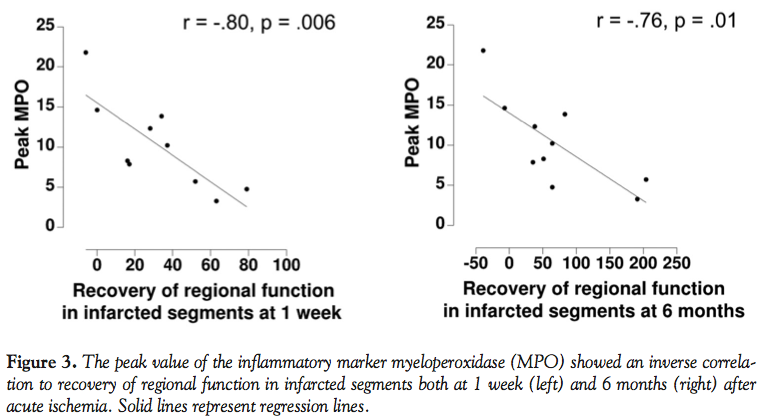 Recovery of regional function. Peak value of myeloperoxidase correlated inversely to recovery of regional function in the infarcted segments between the acute setting and both at 1 week (r = -0.80 and p = 0.006) and at 6 months (r = -0.76 and p = 0.01), as shown in Figure 3. Dysfunctional segments are represented by negative recovery of regional function in Figure 3.
Recovery of regional function. Peak value of myeloperoxidase correlated inversely to recovery of regional function in the infarcted segments between the acute setting and both at 1 week (r = -0.80 and p = 0.006) and at 6 months (r = -0.76 and p = 0.01), as shown in Figure 3. Dysfunctional segments are represented by negative recovery of regional function in Figure 3.
No other statistically significant correlations were found for peak or cumulative markers respectively and recovery of regional function (infarcted segments: |r| < 0.70; adjacent: |r| < 0.70; remote: |r| < 0.55; all p = ns).
Discussion
The main findings were that levels of circulating inflammatory marker neutrophil-gelatinase-associated lipocalin and matrix remodelling marker matrix metalloproteinase-9 have inverse correlations to myocardium at risk. Further, peak myeloperoxidase was found to correlate inversely to salvage and to recovery of regional function over time, and levels of circulating markers for inflammation, matrix remodelling, or reactive oxygen species did not correspond to MI size.
The present study showed no statistically significant correlation between the studied biochemical markers and MI size in man, which is in accordance with previous studies in animals.26 An inverse trend was found, challenging that anti-inflammatory treatment is efficient to reduce MI size in man. This may partly explain the disappointing results from earlier studies where anti-inflammatory treatment in man increased MI size, despite opposite findings in animals.8–14 The weak correlation between the studied biochemical markers and MI size could, however, also be expected since myocardium at risk and duration of ischemia may be considered more important factors for determining final MI size. When studying the inflammatory processes of myocardial ischemia, it may thus be of great importance to study myocardium at risk, rather than MI size. Using biochemical markers acquired before PCI would be a convenient way of measuring myocardium at risk. The results of the present study, however, showed that the studied markers cannot be used for defining myocardium at risk before PCI. One possible explanation for the inverse relation shown between circulating markers and myocardium at risk could be that markers are consumed or accumulated in the affected ischemic myocardial region after reperfusion. This hypothesis and findings from previous experimental studies that have shown an increase of markers of inflammatory response in the ischemic region,21,27 could make it interesting to differentiate between measurements made in ischemic myocardium and in peripheral blood.
A higher degree of salvage was seen with less neutrophil activity in peripheral blood. Increased salvage may be seen as an outcome of smaller final MI size after reperfusion therapy, and thus related to neutrophil activity as proposed by experimental studies.8–10 This indicates the value of further decrease of inflammatory response during reperfusion therapy also in man, despite that no relation was shown between the studied biochemical markers and MI size in itself. It is, however, likely that also salvage is more dependent on duration of ischemia than on neutrophil activation alone. If neutrophil deactivation was crucial to limit MI size, it may be hypothesized that patients with shorter duration of ischemia also would have less neutrophil activation. A correlation between duration of ischemia and neutrophil activation was, however, not seen in the present study. The direct evidence for neutrophil-mediated myocardial cell death thus needs to be further studied in man in relation to myocardium at risk and duration of ischemia.
Stunning has been shown to be attenuated in experimental studies where neutrophils and reactive oxygen species have been downregulated. This has led to the suggestion that neutrophils and reactive oxygen species cause stunning.5 Cytokines also play an important role in the initial ischemic injury,28 as well as in regulation of myocytes and further repairing of the myocardium.29–31 The levels of circulating cytokines interleukin 6 and tumor necrosis factor-α generally increased after reperfusion in the present study. Also, inflammatory response or reactive oxygen species response, measured peripherally, was indicated to have no correlation to regional functional recovery, with the possible exception of myeloperoxidase. This finding is in accordance with previous studies showing that the time course of neutrophil recruitment in stunned myocardium does not indicate that they have a role in stunning.32,33
The results found in the present study may have been highly influenced by the administered GP IIb/IIIa inhibitor, which has previously been proven to attenuate circulating inflammatory markers,34 and to inhibit consequent oxidative reactions.35,36 The administration of heparin may further have decreased the levels of circulating biochemical markers of reactive oxygen species.34
Study limitations. As GP IIb/IIIa inhibitor and heparin were used, these agents may have attenuated circulating markers and thus have impact on the results. Doses administered, however, were according to body weight and it is thus not likely that they were the main cause of the inverse relations found in the present study. It would also have been unethical not to use these agents, as they have been shown to have positive outcomes after reperfusion. The present study had a small sample size, mainly caused by the need of acute myocardial perfusion SPECT to assess myocardium at risk. Myocardium at risk may now be assessed by CMR,37 giving an opportunity to repeat the study in a higher number of patients. Only one female was enrolled, and therefore extrapolation to a female population may not be adequate. Finally, mass significance may have influenced the results. However, since 3 out of 18 parameters for myocardium at risk were found to be statistically significant, mass significance alone cannot explain the findings.
Conclusion
This is the first study in man to show inverse relations between myocardium at risk and levels of circulating markers for inflammation, matrix remodelling, and reactive oxygen species. The current results do not support that reactive oxygen species have a role in stunning in man. Peripheral measurements may not be representative of myocardial inflammatory response and a comparative study between myocardial and peripheral response is needed to evaluate this in man. No significant correlation was found between the studied circulating markers and MI size, challenging the notion that inflammatory treatment is efficient to reduce MI size in man.
Acknowledgements. We gratefully acknowledge technicians Maria Hansson at the Department of Clinical Chemistry, Lund University Hospital, and Barbro Palmqvist at the Wallenberg Laboratory, Malmö University Hospital, and associate professor Lillemor Mattsson-Hultén and professor Olov Wiklund, Wallenberg Laboratory, Gothenburg University, for assistance with analyses. Helen Soneson at the Department of Clinical Physiology, Lund University Hospital, gave invaluable help with MatLab, and statistician Peter Höglund, RSKC, Lund University Hospital, provided expert advice on statistical analyses.
References
- Hedström E, Engblom H, Frogner F, et al. Infarct evolution in man studied in patients with first-time coronary occlusion in comparison to different species — implications for assessment of myocardial salvage. JCMR. 2009;11:38.
- Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Lancet. 1988;2:349-360.
- Braunwald E, Kloner RA. Myocardial reperfusion: a double-edged sword? J Clin Invest. 1985;76:1713-1719.
- Jennings R, Reimer K. Lethal reperfusion injury: fact or fancy? Myocardial Response to Acute Injury. Parratt JR (ed). 1992: pp. 17-34.
- Bolli R. Oxygen-derived free radicals and postischemic myocardial dysfunction (“stunned myocardium”). J Am Coll Cardiol. 1988;12:239-249.
- Bolli R, Zhu WX, Hartley CJ, et al. Attenuation of dysfunction in the postischemic ‘stunned’ myocardium by dimethylthiourea. Circulation. 1987;76:458-468.
- Vinten-Johansen J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc Res. 2004;61:481-497.
- Libby P, Maroko PR, Bloor CM, et al. Reduction of experimental myocardial infarct size by corticosteroid administration. J Clin Invest. 1973;52:599-607.
- Crawford MH, Grover FL, Kolb WP, et al. Complement and neutrophil activation in the pathogenesis of ischemic myocardial injury. Circulation. 1988;78:1449-1458.
- Mullane KM, Read N, Salmon JA, et al. Role of leukocytes in acute myocardial infarction in anesthetized dogs: relationship to myocardial salvage by anti-inflammatory drugs. J Pharmacol Exp Ther. 1984;228:510-522.
- Baran KW, Nguyen M, McKendall GR, et al. Limitation of Myocardial Infarction Following Thrombolysis in Acute Myocardial Infarction (LIMIT AMI) Study Group. Double-blind, randomized trial of an anti-CD18 antibody in conjunction with recombinant tissue plasminogen activator for acute myocardial infarction: limitation of myocardial infarction following thrombolysis in acute myocardial infarction (LIMIT AMI) study. Circulation. 2001;104:2778-2783.
- Dove A. CD18 trials disappoint again. Nat Biotechnol. 2000;18:817-818.
- Roberts R, DeMello V, Sobel BE. Deleterious effects of methylprednisolone in patients with myocardial infarction. Circulation. 1976;53:I204-1206.
- Rusnak JM, Kopecky SL, Clements IP, et al. An anti-CD11/CD18 monoclonal antibody in patients with acute myocardial infarction having percutaneous transluminal coronary angioplasty (the FESTIVAL study). Am J Cardiol. 2001;88:482-487.
- Åström-Olsson K, Hedström E, Hultén LM, et al. Dissociation of the inflammatory reaction following PCI for acute myocardial infarction. J Invasive Cardiol. 2007;19:452-456.
- Bell D, Jackson M, Nicoll JJ, et al. Inflammatory response, neutrophil activation, and free radical production after acute myocardial infarction: effect of thrombolytic treatment. Br Heart J. 1990;63:82-87.
- Lalu MM, Pasini E, Schulze CJ, et al. Ischaemia-reperfusion injury activates matrix metalloproteinases in the human heart. Eur Heart J. 2005;26:27-35.
- Hemdahl AL, Gabrielsen A, Zhu C, et al. Expression of neutrophil gelatinase-associated lipocalin in atherosclerosis and myocardial infarction. Arterioscler Thromb Vasc Biol. 2006;26:136-142.
- Puhakka M, Magga J, Hietakorpi S, et al. Interleukin-6 and tumor necrosis factor alpha in relation to myocardial infarct size and collagen formation. J Card Fail. 2003;9:325-332.
- Smit JJ, Ottervanger JP, Slingerland RJ, et al. On-TIME Study Group. Comparison of usefulness of C-reactive protein versus white blood cell count to predict outcome after primary percutaneous coronary intervention for ST elevation myocardial infarction. Am J Cardiol. 2008;101:446-451.
- Lu L, Gunja-Smith Z, Woessner JF, et al. Matrix metalloproteinases and collagen ultrastructure in moderate myocardial ischemia and reperfusion in vivo. Am J Physiol/Heart Circ Physiol. 2000;279:H601-609.
- Engblom H, Hedström E, Heiberg E, et al. Rapid initial reduction of hyperenhanced myocardium after reperfused first myocardial infarction suggests recovery of the peri-infarction zone: one-year follow-up by MRI. Circ Cardiovasc Imaging. 2009;2:47-55.
- Soneson H, Ubachs JF, Ugander M, et al. An improved method for automatic segmentation of the left ventricle in myocardial perfusion SPECT. J Nucl Med. 2009;50:205-213.
- Soneson H, Engblom H, Hedström E, et al. An automatic method for quantification of myocardium at risk from myocardial perfusion SPECT in patients with acute coronary occlusion. J Nucl Cardiol. 2010;17:831-840.
- Heiberg E, Ugander M, Engblom H, et al. Automated quantification of myocardial infarction from MR images by accounting for partial volume effects: animal, phantom, and human study. Radiology. 2008;246:581-588.
- Baxter GF. The neutrophil as a mediator of myocardial ischemia-reperfusion injury: time to move on. Basic Res Cardiol. 2002;97:268-275.
- Mullane KM, Kraemer R, Smith B. Myeloperoxidase activity as a quantitative assessment of neutrophil infiltration into ischemic myocardium. J Pharmacol Methods. 1985;14:157-167.
- Gwechenberger M, Mendoza LH, Youker KA, et al. Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation. 1999;99:546-551.
- Gallucci RM, Simeonova PP, Matheson JM, et al. Impaired cutaneous wound healing in interleukin-6-deficient and immunosuppressed mice. FASEB J. 2000;14:2525-2531.
- Finkel MS, Oddis CV, Jacob TD, et al. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science 1992;257:387-389.
- Jordan JE, Zhao ZQ, Vinten-Johansen J. The role of neutrophils in myocardial ischemia-reperfusion injury. Cardiovasc Res. 1999;43:860-878.
- Go LO, Murry CE, Richard VJ, et al. Myocardial neutrophil accumulation during reperfusion after reversible or irreversible ischemic injury. Am J Physiol. 1988;255:H1188-H1198.
- Juneau CF, Ito BR, del Balzo U, et al. Severe neutrophil depletion by leucocyte filters or cytotoxic drug does not improve recovery of contractile function in stunned porcine myocardium. Cardiovasc Res. 1993;27:720-727.
- Karlsson K, Marklund SL. Heparin-induced release of extracellular superoxide dismutase to human blood plasma. Biochem J. 1987;242:55-59.
- Lincoff AM, Kereiakes DJ, Mascelli MA, et al. Abciximab suppresses the rise in levels of circulating inflammatory markers after percutaneous coronary revascularization. Circulation. 2001;104:163-167.
- Neumann FJ, Kastrati A, Schmitt C, et al. Effect of glycoprotein IIb/IIIa receptor blockade on platelet-leukocyte interaction and surface expression of the leukocyte integrin Mac-1 in acute myocardial infarction. J Am Coll Cardiol. 1999;34:1420-1426.
- Carlsson M, Ubachs JF, Hedström E, et al. Myocardium at risk after acute infarction in humans on cardiac magnetic resonance: quantitative assessment during follow-up and validation with single-photon emission computed tomography. JACC Cardiovasc Imaging. 2009;2:569-576.
__________________________________
From the 1Department of Clinical Physiology, 2Department of Cardiology, and the 3Department of Laboratory Medicine, Lund University and Lund University Hospital, Lund, Sweden.
Disclosure: The authors have completed and returned the ICMJE Form for Disclosure of Potential Conflicts of Interest. No authors reported conflicts regarding the content herein.
Funding: The study was supported by grants from the region of Skane county coun- cil’s research and development foundation, the Ernhold Lundström Foundation, Research Funds at University Hospital MAS, the Hulda Almroth Foundation, the Medical Faculty at Lund University, Sweden Research Council (521-2008-2461), and the Swedish Heart and Lung Foundation.
Manuscript submitted April 4, 2011, provisional acceptance given May 11, 2011, final version accepted July 25, 2011.
Address for correspondence: Professor Håkan Arheden, Department of Clinical Physiology, SE-22185 LUND, Sweden. Email: hakan.arheden@med.lu.se