


Local Delivery of Gene-Based Therapy for Hepatocellular Carcinoma: The TACE of the Future?
From the 1Department of Biomedical Engineering, and the Translational Tissue Engineering Center, Johns Hopkins University, Baltimore, Maryland, and the Instituto do Câncer do Estado de São Paulo, Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, São Paulo, Brazil; 2Departments of Biomedical Engineering, Neurosurgery and Ophthalmology, and the Translational Tissue Engineering Center, Johns Hopkins University School of Medicine and Department of Materials Science and Engineering, Johns Hopkins University, Baltimore, Maryland; and 3Russell H. Morgan Department of Radiology and Radiological Science, Johns Hopkins Medical Institutions, Baltimore, Maryland.
Abstract: Hepatocellular carcinoma (HCC) accounts for approximately 90% of primary liver cancers and has a 5-year postdiagnosis survival rate of only 12%. This poor prognosis is due to the fact that HCC has often spread throughout the liver parenchyma or metastasized elsewhere at the time of diagnosis. Unfortunately, conventional therapy has limited efficacy for these cases. Transarterial chemoembolization (TACE) is a minimally invasive procedure increasingly utilized for invasive HCC palliation. While there are several advantages related to the hepatic artery delivery approach, such as reduced systemic toxicity, TACE is not curative and has several limitations including HCC chemoresistance and regional toxicity. TACE often damages surrounding healthy hepatocytes, which can lead to liver failure and associated morbidity. Recent research efforts aimed at developing novel nonviral vectors are encouraging and suggest that the concept of cancer gene therapy may yet prove to be a safe and effective treatment option for HCC patients. Non-viral gene-based treatments can be engineered to prevent off-target effects and promote cancer-specific therapy on the molecular and cellular levels. Here the pharmacokinetics of transarterial delivery from which TACE benefits are reviewed and a possible successful marriage between the hepatic artery delivery approach and gene therapy for HCC treatment is discussed.
Key words: genetic therapy, drug administration routes, chemoembolization, interventional oncology
_______________________________________
Liver cancer has the sixth highest cancer incidence and is the second leading cause of cancer mortality worldwide.1 Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer, accounting for up to 90% of cases.2 While HCC is more common in developing countries, the incidence in the United States has tripled in the past 30 years and continues to rise.3,4 Currently, HCC is the third most common cause of cancer death in the United States and has the fastest-growing mortality rates among all cancers. The overall 5-year survival for HCC patients is estimated to be a meager 12%.3,4 HCC is often invasive and in 70% of cases the disease has spread throughout the liver parenchyma or metastasized elsewhere at the time of diagnosis.5,6 Patients with invasive HCC are ineligible for curative treatments.7,8
Transarterial chemoembolization (TACE) is a palliative approach that is currently considered the standard of care for asymptomatic patients with multiple liver tumors and no vascular or extrahepatic invasion.6-8 This minimally invasive procedure is based on the local delivery of chemotherapeutic and embolization agents via the hepatic artery and its branches.9 The local approach of the hepatic artery takes advantage of the predominant arterial supply to intrahepatic HCC lesions, enabling selective embolization of the tumor feeding vessels.10,11 Local delivery can afford reduced systemic exposure to anticancer drugs and thus lower toxicity relative to systemic administration.12
Unfortunately, TACE has significant limitations. Many anticancer drugs, such as etoposide13 and methotrexate,14 have unfavorable pharmacokinetics for local delivery and, in such cases, local administration cannot mitigate systemic toxicity.12 Additionally, while TACE has proven effective for palliation, recurrence is expected, because HCC tumors develop or have inherent chemoresistance, limiting the long-term efficacy of treatment.15 Moreover, juxtaposed healthy hepatocytes remain vulnerable to embolization and chemotherapeutic toxicity in spite of the relative regioselectivity that TACE affords. Liver failure caused by the injury to normal tissue is a common complication and the main cause of mortality among patients treated with TACE.16,17
Gene therapy offers promising advantages that might overcome the limitations and undesired effects of TACE. Unlike conventional therapy, gene therapy has the potential to target cancer cells while sparing healthy tissue.18 Recent research seeks to elucidate the mechanisms governing gene delivery and molecularly targeted gene expression, enabling development of multiple cancer targeting strategies.19 Given the pharmacokinetics of gene-based treatments, local delivery of nucleic acid based therapies may prove to be the TACE of the future. Herein we aim to review the pharmacokinetics governing hepatic artery delivery and demonstrate the potential advantages of gene therapy for the treatment of HCC.
Pharmacokinetics and Local Delivery Advantage
Most drugs need to undergo liver metabolism before being eliminated. The loss of a drug across the liver, or the hepatic clearance (CLH), is often partially or totally responsible for drug elimination from the body (total body clearance or CLTB). To get cleared by the liver, a drug must enter the hepatic circulation and be extracted by the hepatocytes. Thus, the hepatic clearance depends on the blood flow entering the liver (QH) and the hepatic extraction ratio (EH) (Equation 1).20
Equation 1. CH=QH*EH
When the liver is the target site for drug-based treatment, the local delivery of drugs with hepatic metabolism can potentially reduce the systemic toxicity by enabling the elimination of the drug before entering the systemic circulation.21 In these cases, however, the advantages of local over systemic delivery are highly dependent on the drug extracted in the first passage through the liver: the higher the hepatic extraction ratio in the first passage, the lower the systemic exposure and off-target effects.22,23 This reduction in systemic toxicity also enables higher doses to be delivered locally than systemically, resulting in improved tumor response, especially for drugs with steep dose-response curves.12,24 In contrast, drugs without hepatic metabolism do not experience first-pass effect and, thus, the systemic toxicity will not be reduced by the choice of the local route. In these cases, the possible advantage for local delivery will depend on the increase in concentration in the target site that can be achieved if the same dose is given locally to the liver compared to systemically.12,24 This rationale enables the prediction of the improvement in concentration that can be achieved if local injection is preferred over systemic administration, or the advantage of local over systemic delivery (Rd) (Equation 2).12,24,25
According to pharmacodynamics principles (dose-response relationship), typical drugs with a sigmoidal dose-response curve demonstrate improved efficacy when the concentration in the target tissue is increased along the steep portion of the curve. Therefore, higher concentrations are desirable.12,22
Equation 2. Rd=1+CLTB/QH(1–EH)
According to Equation 2, the delivery of a drug that has a high hepatic extraction ratio and is rapidly cleared by the body (high CLTB) through a route with low exchange rate (low Q) provides the best advantage for local delivery.25
Assuming that the liver can be considered the only metabolizing tissue of many drugs,26,27 equation 2 can be simplified to Equation 3.12
Equation 3. Rd=1/(1–EH)
In this cases, the drug concentration achieved in the target site after local delivery is virtually the same as the concentration that follows systemic injection of the same dose. However, the higher the hepatic extraction ratio, the higher the decrease in systemic toxicity, and thus, the local dose can be raised for local delivery.12
Pharmacokinetics: TACE and DEB-TACE
TACE and other analogous procedures such as drug-eluting bead TACE (DEB-TACE) function by promoting chemical and mechanical-mediated cancer cell death. The embolization of tumor-feeding branches of the hepatic artery causes a regioselective ischemia of the malignant tissue. The ischemia promotes cell death through necrosis, which results in tumor regression. The embolization process also causes reduction of the local exchange rate,25 which drops from relatively high hepatic artery flows of 633 mL/min on average for patients with intrahepatic metastasis28 to values close to zero in cases of complete blockage.29 The embolization of the hepatic artery and/or its branches combined with the delivery of drugs with high hepatic extraction ratios, high CLTB (Equation 2), and a steep dose-response curve can maximize the advantage of local delivery.25,30
Floruxidine (5-fluoro-2′-deoxyuridine; FUDR) is a chemotherapeutic drug mainly used in the treatment of hepatic metastasis of colorectal cancer.23,27 It has pharmacokinetic and pharmacodynamic properties extremely favorable for local delivery to the liver. FUDR has a steep dose-response curve31 and high hepatic extraction ratio, lying in the range of 0.69 to 0.92, with 94% to 99% of the drug being extracted in the first pass.22 Also, FUDR has a short systemic half-life. The hepatic artery-to-systemic concentration ratio (Rd) for of FUDR has been described to be as high as 400.25
The most commonly utilized drug for transarterial treatment of HCC is doxorubicin32 either as a monotherapy or in combination with other drugs.33-35 Doxorubicin is currently administered mixed with lipiodol (TACE) or as doxorubicin-loaded drug-eluting beads (DEB-TACE).36 Doxorubicin is an anthracycline antibiotic that has anticancer activity to diverse cancer types.37,38 Doxorubicin therapeutic effects occur mainly following apoptosis-mediated cell death, but also through autophagy and necrosis. Free radicals are generated by doxorubicin redox cycling, which can cause major toxic effects,30,36 most notably irreversible cardiotoxicity characterized by dilated cardiomyopathy and congestive heart failure.32,38,39 The high risk of cardiomyopathy usually limits the use of doxorubicin to a cumulative dose of 550 mg/m2;40 however, some patients develop this complication after a single dose.41 The main doxorubicin metabolite, doxorubicinol, also presents systemic toxicity.42,43 Since the hepatic extraction ratio of doxorubicin is only 0.45 to 0.5,44 a hepatic artery administration still leads to systemic exposure and doxorubicinol prolongs systemic toxicity even after the hepatic metabolism of doxorubicin. Hepatocyte damage is also an undesirable doxorubicin side effect mediated by free-radical production. In a prospective study, Damodar et al observed hepatotoxicity in 30.4% of patients receiving this drug.45 HCC recurrence after TACE has occurred due to doxorubicin resistance of liver cancer stem cell (CSC) populations. Cancer stem cells are responsible for aggressive HCC phenotypes including chemoresistance and metastatic potential.46 In general, hypoxia has been suggested to be a significant cause of chemoresistance, which is particularly critical in the context of HCC, since TACE induces acute hypoxia.15 Even though complete or partial tumor regression is reported to occur post-TACE with doxorubicin, these results are mainly temporary47 and the advantage of TACE over transarterial embolization (TAE) alone remains a controversial question.48-50
TACE and DEB-TACE are two procedures commonly used in the palliation of patients with multiple intrahepatic HCC metastases. While in conventional TACE, intra-arterial injection of doxorubicin-lipiodol is followed by administration of embolization agents, in DEB-TACE the beads (microspheres) work as both a slow-release drug carrier and an embolization agent. Hong et al have evaluated the pharmacokinetic differences between these two techniques using a rabbit HCC model. The authors observed a statistically significant decrease in the peak plasma concentration of doxorubicin when administered with DEB-TACE in comparison to intra-arterial (IA) injection of drug alone (i.e., no embolic agent). In this rabbit model, no statistically significant difference was observed between DEB-TACE and TACE. Following DEB-TACE, steady increase in plasma doxorubicinol was combined with a decrease in plasma doxorubicin levels and doxorubicin could be detected in the tumor tissue at 14 days posttreatment, indicative of the slow drug release mechanism of the DEB within the tumor.51 In humans, Varela et al showed that the peak plasma concentration and the AUC (area under the curve) of plasma doxorubicin were significantly lower among DEB-TACE patients when compared to the TACE cohort, even though the mean doxorubicin dose administered was higher in DEB-TACE.52 Regarding the therapeutic effect, a recent meta-analysis by Huang et al argued that DEB-TACE achieves a better tumor response as compared to conventional TACE.53 However, Gao et al54 and Golfieri et al47 found no difference in therapeutic efficacy between these two treatment options in a meta-analysis and a randomized clinical trial, respectively. In short, large randomized controlled studies are required to definitively establish the superiority of DEB-TACE relative to TACE.
Gene Therapy
Gene therapy consists of the transfer of genetic materials to cells of interest to achieve therapeutic effects.55 The efficacy of a gene-based treatment depends on several factors such as route of administration, choice of the nucleic acid, delivery vector, and gene expression.56,57 While plasmid DNAs containing therapeutic genes can be used to introduce or reintroduce lacking, lost, or mutated proteins to ultimately promote cancer regression,58,59 small interfering RNAs (siRNAs) are delivered to cancer cells to silence genes involved in the oncogenic process.60-62 Currently, the poor selectivity in vector targeting, ineffective gene delivery, and safety issues represent the most critical barriers preventing gene therapy to succeed as a powerful and viable treatment option for cancer and other genetic-based diseases.63
Viral and nonviral vectors have been used to deliver recombinant nucleic acids. Viruses have the natural capacity to promote effective infection of the host cells, overcome intracellular barriers, and transfer viral genetic material to the nucleus.64 However, the widespread use and clinical translation of viral vectors have been limited by its costly production,65 potential immunogenicity, and tumorigenicity from insertional mutagenesis.66,67 In addition, the membrane receptors that mediate virus internalization are not specific for cancer cells and the attachment of cancer-targeting ligands to the viral capsid can lead to reduced transduction efficacy.68 The vast majority of researchers in gene therapy for HCC have been using viral carriers, because nonviral vectors are relatively new.
Lipid and polymeric-based nonviral vectors are capable to complex with nucleic acids and form nanoparticles that can be uptaken by the cell.67,69 Such vectors are engineered to allow favorable intracellular trafficking in order for the nucleic acid to reach its final location, for example, cytoplasm for RNA and nucleus for DNA. Nonviral vectors are generally nonimmunogenic, less expensive to produce, and able to carry larger amounts of nucleic acid.67,70-72 While lipid-based vectors can have high transfection efficacy and important nonspecific cytotoxic effects, cationic polymers can be biodegradable and safer but are often considered to promote inferior gene-delivery results.67
Polymeric biomaterials are promising gene delivery vectors that have potential to be easily modified. They can be tailored to optimize transfection efficacy and to promote cancer-targeting and cancer-specific delivery.19,67 Cationic polymers self-assemble with nucleic acids, which are anionic, through electrostatic interactions.67,69 Resulting polyplex (nanoparticles) have biophysical and biomaterial-mediated cancer-targeting properties.19 Nanoparticles smaller than 500 nm can accumulate at the tumor site due to the permeable vessels and insufficient lymphatic drainage, commonly present in solid tumors (enhanced permeation and retention effect).73 The positively charged particles interact with negative charges on the cell surface and are uptaken by the cell.74,75 Polymeric nanoparticles can also be conjugated to select ligands whose receptors are usually upregulated in cancer tissues.19,67 Additionally, structure-dependent tissue-specific gene delivery has been observed.76-80 Further cancer specificity through gene therapy can also be achieved at the transcriptional (Figure 1), post-transcriptional, and translational levels.19
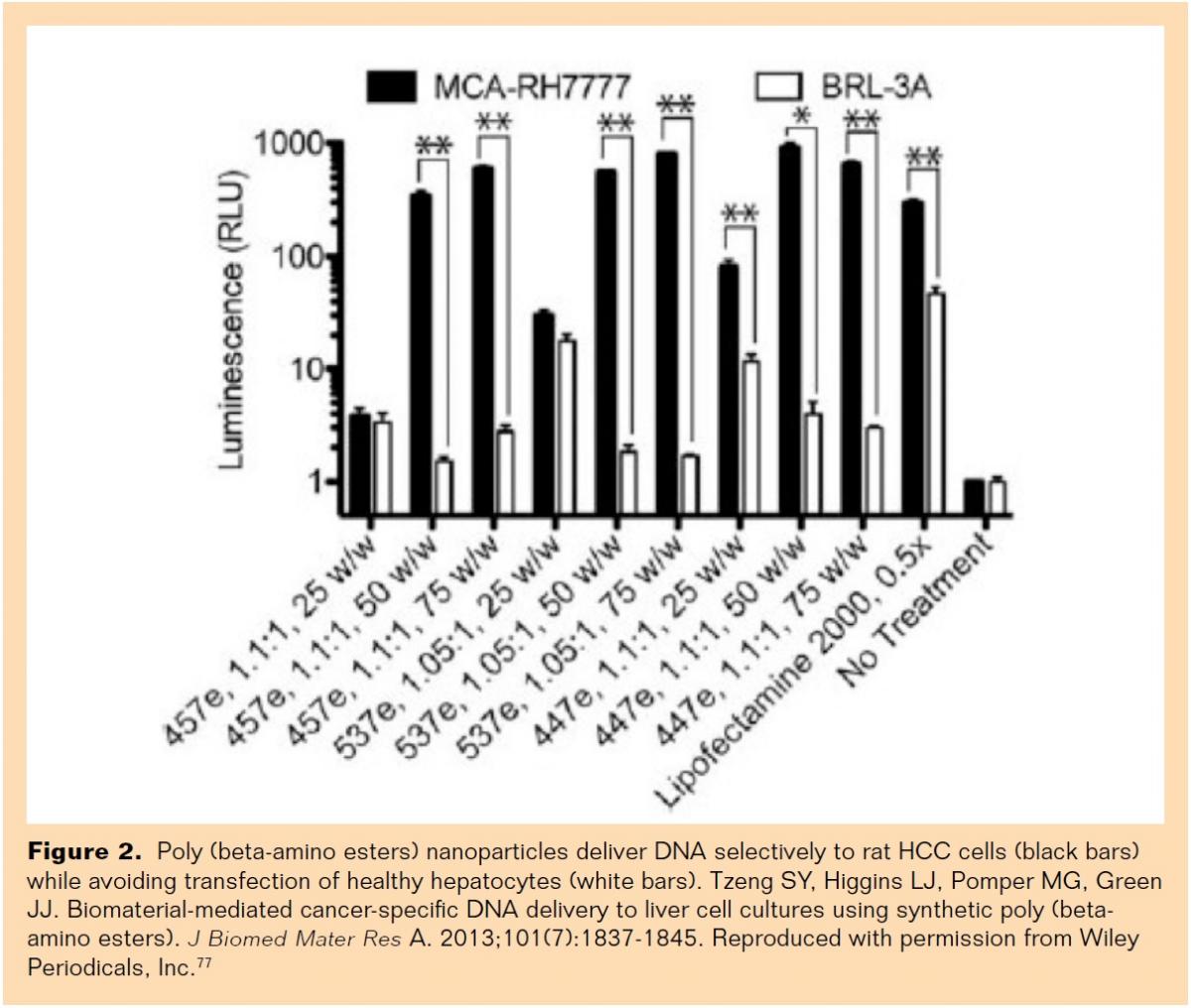 Poly (beta-amino esters) (PBAEs) are cationic polymers that, unlike other commonly used polymers such as polyethylenimine (PEI), can be degraded by hydrolysis (biodegradable), which allows the release of nucleic acid inside the cytoplasm and reduces cytotoxicity.81 PBAE nanoparticles have also shown to present intrinsic biomaterial-mediated cell specificity, delivering DNA to cancer cells while avoiding healthy tissue.76,77 Tzeng et al recently demonstrated that specific PBAE polymer structures are able to deliver reporter genes to a rat HCC cell line (MCA-RH7777) with high efficacy and remarkable specificity over rat hepatocytes (BRL-3A) in vitro (Figure 2).77
Poly (beta-amino esters) (PBAEs) are cationic polymers that, unlike other commonly used polymers such as polyethylenimine (PEI), can be degraded by hydrolysis (biodegradable), which allows the release of nucleic acid inside the cytoplasm and reduces cytotoxicity.81 PBAE nanoparticles have also shown to present intrinsic biomaterial-mediated cell specificity, delivering DNA to cancer cells while avoiding healthy tissue.76,77 Tzeng et al recently demonstrated that specific PBAE polymer structures are able to deliver reporter genes to a rat HCC cell line (MCA-RH7777) with high efficacy and remarkable specificity over rat hepatocytes (BRL-3A) in vitro (Figure 2).77
Gene therapy has been mainly applied to promote apoptosis-mediated cancer cell death and to inhibit tumor progression.18 Cell death through apoptosis is desirable, since it is generally not followed by inflammation that results in the damage of surrounding tissue, contrarily to the necrosis-mediated cell death that accompanies the embolization procedure on TACE. Additionally, hypoxia that follows embolization promotes hepatocarcinogenesis by enhancing cell proliferation and inducing angiogenesis, metastasis, and chemoresistance.82 The tumor suppressor gene p53 is involved in both intrinsic and extrinsic apoptotic pathways83 and its function is often impaired in HCC.84-88 It has been demonstrated that the reintroduction of the wild-type p53 to HCC cells can restore apoptotic capacity, inhibit tumor progression89,90 and induce sensitization to chemotherapy.91 On the other hand, RNA interference has shown successful results in knockdown oncogenes overexpressed in HCC, such as beta-catenin,92 survivin,93 and human gankyrin gene product (p28GANK).94 Zhang et al demonstrated tumor growth suppression due to increased apoptotic susceptibility and prolonged survival in HCC mice xenographs following siRNA-mediated silencing of the survivin gene.93 HCC cell death can also be induced by delivering genes capable of activating the extrinsic apoptotic pathway, such as the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) gene,95 and the so-called suicide genes,96 which are capable of promoting conversion of prodrug in active cell-killing agents.97 The choice of select genes, such as the ones referenced above, enable an extra level of tumor-directed treatment.19 Overexpression of oncogenes and mutation or loss of tumor suppressor genes are intrinsic of the tumorigenic process, TRAIL is cancer-selective,98 and the HSVtk/gancivlovir system has a bystander effect that allows specific cell killing of nontransfected rapid dividing cells.99 Additionally, the specificity of gene expression in cancer cells can be considerably increased by cancer-specific promoters.100 During the HCC tumorigenesis, α-fetoprotein (AFP), a fetal protein, often becomes re-expressed. The promoter of the AFP gene is, therefore, considered HCC-specific101 and has been demonstrated to be a powerful ally in gene therapy for HCC. Hu et al, for example, showed that the AFP promoter enables cell killing mediated by the tBid gene overexpression only in AFP-producing HCC.102 The hepatocarcinoma-intestine-pancreas gene promoter represents another example of an HCC-specific promoter.103
Local routes of delivery have been successfully applied to further improve the therapy results for HCC. Mohr et al utilized cationic liposomes to deliver a reporter gene to orthotopic human HCC in athymic mice models. The authors found that local (intratumoral) injection of liposome-DNA polyplex provided high levels of gene expression in HCC cells while only minimal expression occurred in surrounding normal liver. Even though the authors have not performed a direct comparison among routes of delivery, they observed that gene expression after tail vein injection was low and not selective for the HCC tumor. Portal vein injection led to high transfection levels to the normal liver and major hepatocyte toxicity.104 In another study, Ahn et al observed that intratumoral administration increased tumor over normal tissue expression in a orthotopic rat model of HCC treated with Ad-pSurv-TSTA-TRAIL-FL, a bidirectional, 2-step transcriptional amplification (TSTA) system driven by survivin, a tumor-specific promoter, to increase expression of both the reporter gene firefly luciferase and the therapeutic gene TRAIL.105 Utilizing VX2 rabbit HCC models, Luo et al compared intratumoral, intra-arterial alone, and intra-arterial plus TAE as administration strategies for rAd-p53 delivery. The authors demonstrated that tumor volumes were significantly smaller in the intra-arterial plus TAE group compared to all the other groups.106-107 Using a diethylnitrosoamine-induced HCC rat model, Bilbao et al found that adenoviral-mediated transduction of a reporter gene via hepatic artery delivery might be related to tumor size and histology, and bigger tumors might benefit from vasoactive drug administration prior to gene therapy.108 Kim et al utilized PEI, a nondegradable cationic polymer, to deliver the reporter gene luciferase to VX2 rabbit HCC models via hepatic artery. The authors demonstrated that the formulation of PEI-luciferase complex mixed with iopamidol and iodized oil led to significantly greater luciferase activity in HCC tumors.107
Clinical Trials
Gene therapy has been increasingly applied in clinical trials for cancer worldwide. Viruses were the first vectors tested clinically for gene therapy and represent the most commonly tested vectors in clinical trials. In 2003, a recombinant adenovirus expressing human p53 gene (rAd-p53) was the first gene therapy product approved by a government for commercialization; it occurred in China and the product received the brand name Gendicine.109 Recently, Chen et al carried out a randomized clinical trial involving 30 patients with intermediate or advanced stage HCC, according to the BCLC classification,8 comparing hepatic artery delivery of chemotherapeutic drug (hydroxycamptothecin) plus Gendicine (treatment group) and hepatic artery chemotherapeutic drug alone (control group) once a week for 3 weeks. The survival rates of the treatment group were significantly higher than the control group at 1, 3, 6, 9, and 12 months post intervention. Besides, the mean rate of mutant p53 significantly decreased in the treatment group when comparing pre and posttreatment (23.74% vs 11.81%, respectively, P<.01).110 In 2009, Tian et al reported the results from 46 patients that were treated with TACE alone or TACE followed by multiple hepatic artery injections of rAd-p53 plus chemotherapy (5-fluorouracil). The authors found no difference between the treatment and the control groups regarding tumor response (69.5% vs 65.2%, respectively), time to progression (9.6 months vs 8.3 months, respectively) and overall survival (12.8% vs 10.4%, respectively).111 Three other trials112-114 compared viral-mediated gene therapy plus TACE versus TACE alone for HCC treatment. All found improved overall survival in the treatment group; however, these trials are retrospective and/or nonrandomized. No major complications were reported by the authors of any of these trials (Table 1). However, severe adverse events induced by viral gene therapy have occurred in other situations. In a phase I clinical trial for gene-based treatment for a metabolic disease, one death followed a severe immune reaction to the adenovirus vector used.115 Additionally, retroviral gene therapy caused insertional mutagenesis and induced leukemia-like disorder in 5 of 20 patients being treated for X-linked severe combined immunodeficiency, resulting in 1 death.116,117 Two clinical trials are currently recruiting patients to evaluate TACE combined with gene therapy for HCC, both applying viral vectors.118,119 Unfortunately, there are no reported or ongoing clinical trials in nonviral gene delivery for HCC or comparing hepatic artery administration to other routes of delivery for gene therapy.
Conclusion and Future Prospects
The future of cancer treatment will not rely on a single therapeutic approach, but instead will be a combination of powerful strategies that together will lead to cancer remission, reduced systemic toxicity, and improved quality of life and survival with personalized medical care. This article summarized the pros and cons of the current management of HCC and the potential for gene therapy incorporation in the transarterial strategy. Many questions still remain unsolved and more studies and clinical trials in nonviral gene therapy for HCC are necessary. Future prospects include further investigation and comparison between local and systemic gene therapy as well as between TACE and gene therapy.
Editor’s note: Disclosure: The authors have completed and returned the ICMJE Form for Disclosure of Potential Conflicts of Interest. The authors report no related disclosures.
Manuscript received June 22, 2015; manuscript accepted August 19, 2015.
Suggested citation: Zamboni CG, Green JJ, Higgins LJ. Local delivery of gene-based therapy for hepatocellular carcinoma: the TACE of the future? Intervent Oncol 360. 2015;3(11):E121-E136.
References
1. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359-E386.
2. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557-2576.
3. El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118-1127.
4. Ries LAG, Eisner MP, Kosary CL, Hankey BF, Miller BA, Clegg L. Surveillance, Epidemiology, and End Results (SEER) Program SEER* Stat Database: Incidence— SEER 9 Regs Public-Use, Nov 2004 Sub (1973–2002). National Cancer Institute, Division of Cancer Control and Population Sciences, Surveillance Research Program, Cancer Statistics Branch. Released April 2005, based on the November 2004 submission. 2008.
5. Llovet JM, Di Bisceglie AM, Bruix J, et al. Design and endpoints of clinical trials in hepatocellular carcinoma. J Natl Cancer Inst. 2008;100(10):698-711.
6. Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362(9399):1907-1917.
7. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379(9822):1245-1255.
8. Llovet JM, Brú C, Bruix J. Prognosis of hepatocellular carcinoma: the BCLC staging classification. Semin Liver Dis. 1999;19(3):329-338.
9. Vogl TJ, Naguib NN, Nour-Eldin NE, et al. Review on transarterial chemoembolization in hepatocellular carcinoma: palliative, combined, neoadjuvant, bridging, and symptomatic indications. Eur J Radiol. 2009;72(3):505-516.
10. Bierman HR, Byron RL, Kelley KH, Grady A. Studies on the blood supply of tumors in man. III. Vascular patterns of the liver by hepatic arteriography in vivo. J Natl Cancer Inst. 1951;12(1):107-131.
11. Breedis C, Young G. The blood supply of neoplasms in the liver. Am J Pathol. 1954;30(5):969-977.
12. Collins JM. Pharmacologic rationale for regional drug delivery. J Clin Oncol. 1984;2(5):498-504.
13. van Tellingen O, Kuck MT, Vlasveld LT, Rodenhuis S, Nooijen WJ, Beijnen JH. Unchanged pharmacokinetics of etoposide given by intra-arterial hepatic infusion as compared with i.v. infusion. Cancer Chemother Pharmacol. 1996;38(4):387-390.
14. Parasrampuria R, Mehvar R. Dose‐dependent inhibition of transporter‐mediated hepatic uptake and biliary excretion of methotrexate by cyclosporine A in an isolated perfused rat liver model. J Pharm Sci. 2010;99(12):5060-5069.
15. Asghar U, Meyer T. Are there opportunities for chemotherapy in the treatment of hepatocellular cancer? J Hepatol. 2012;56(3):686-695.
16. Huang YS, Chiang JH, Wu JC, Chang FY, Lee SD. Risk of hepatic failure after transcatheter arterial chemoembolization for hepatocellular carcinoma: predictive value of the monoethylglycinexylidide test. Am J Gastroenterol. 2002;97(5):1223-1227.
17. Groupe d’Etude et de Traitement du Carcinome Hépatocellulaire. A comparison of lipiodol chemoembolization and conservative treatment for unresectable hepatocellular carcinoma. N Engl J Med. 1995;332(19):1256-1261.
18. Avila MA, Berasain C, Sangro B, Prieto J. New therapies for hepatocellular carcinoma. Oncogene. 2006;25(27):3866-3884.
19. Kim J, Wilson DR, Zamboni CG, Green JJ. Targeted polymeric nanoparticles for cancer gene therapy. J Drug Target. 2015;23(7-8):627-641.
20. Rowland M, Tozer TN. Clinical Pharmacokinetics: concepts and applications, vol 3, 3rd ed. Philadelphia, Pennsylvania: Lippincott Williams & Wilkins, 1995.
21. Ensminger WD, Gyves JW. Clinical pharmacology of hepatic arterial chemotherapy. Semin Oncol. 1983;10(2):176-182.
22. Ensminger WD, Rosowsky A, Raso V, et al. A clinical-pharmacological evaluation of hepatic arterial infusions of 5-fluoro-2′-deoxyuridine and 5-fluorouracil. Cancer Res. 1978;38(11 Part 1):3784-3792.
23. Power DG, Kemeny NE. The role of floxuridine in metastatic liver disease. Mol Cancer Ther. 2009;8(5):1015-1025.
24. Chen HS, Gross JF. Intra-arterial infusion of anticancer drugs: theoretic aspects of drug delivery and review of responses. Cancer Treat Rep. 1980;64(1):31-40.
25. Ensminger WD, Gyves JW. Regional chemotherapy of neoplastic diseases. Pharmacol Ther. 1983;21(2):277-293.
26. Lerapetritou MG, Georgopoulos PG, Roth CM, Androulakis LP. Tissue‐level modeling of xenobiotic metabolism in liver: an emerging tool for enabling clinical translational research. Clin Transl Sci. 2009;2(3):228-237.
27. Sugarbaker PH, Mora JT, Carmignani P, Stuart OA, Yoo D. Update on chemotherapeutic agents utilized for perioperative intraperitoneal chemotherapy. Oncologist. 2005;10(2):112-122.
28. Leen E, Goldberg JA, Anderson JR, et al. Hepatic perfusion changes in patients with liver metastases: comparison with those patients with cirrhosis. Gut. 1993;34(4):554-557.
29. Lencioni R, Petruzzi P, Crocetti L. Chemoembolization of hepatocellular carcinoma. Semin Intervent Radiol. 2013;30(1):3-11.
30. Cohen AD, Kemeny NE. An update on hepatic arterial infusion chemotherapy for colorectal cancer. Oncologist. 2003;8(6):553-566.
31. Creaven PJ, Rustum YM, Petrelli NJ, et al. Phase I and pharmacokinetic evaluation of floxuridine/leucovorin given on the Roswell Park weekly regimen. Cancer Chemother Pharmacol. 1994;34(3):261-265.
32. Tam K. The roles of doxorubicin in hepatocellular carcinoma. ADMET and DMPK. 2013;1(3):29-44.
33. Nishikawa H, Osaki Y, Kita R, Kimura T. Hepatic arterial infusion chemotherapy for advanced hepatocellular carcinoma in Japan. Cancers (Basel). 2012;4(1):165-183.
34. Bruix J, Sala M, Llovet JM. Chemoembolization for hepatocellular carcinoma. Gastroenterology. 2004;127(5):S179-S188.
35. Basile A, Carrafiello G, Ierardi AM, Tsetis D, Brountzos E. Quality-improvement guidelines for hepatic transarterial chemoembolization. Cardiovasc Intervent Radiol. 2012;35(4):765-774.
36. Liapi E, Geschwind JF. Transcatheter arterial chemoembolization for liver cancer: is it time to distinguish conventional from drug-eluting chemoembolization? Cardiovasc Intervent Radiol. 2011;34(1):37-49.
37. Rahman AM, Yusuf SW, Ewer MS. Anthracycline-induced cardiotoxicity and the cardiac-sparing effect of liposomal formulation. Int J Nanomedicine. 2007;2(4):567-583.
38. Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65(2):157-170.
39. Mozdzanowska D, Woźniewski M. Radiotherapy and anthracyclines-cardiovascular toxicity. Contemp Oncol (Pozn). 2015;19(2):93-97.
40. Von Hoff DD, Layard MW, Basa P, et al. Risk factors for doxorubicin-lnduced congestive heart failure. Ann Intern Med. 1979;91(5):710-717.
41. Devoy MA, Tomson CR. Fatal cardiac failure after a single dose of doxorubicin in myeloma-associated cardiac amyloid. Postgrad Med J. 1992;68(795):69-69.
42. Dessypris EN, Brenner DE, Baer MR, Hande KR. Uptake and intracellular distribution of doxorubicin metabolites in B-lymphocytes of chronic lymphocytic leukemia. Cancer Res. 1988;48(3):503-506.
43. Olson RD, Mushlin PS, Brenner DE, et al. Doxorubicin cardiotoxicity may be caused by its metabolite, doxorubicinol. Proc Natl Acad Sci U S A. 1988;85(10):3585-3589.
44. Garnick MB, Ensminger WD, Israel M. A clinical-pharmacological evaluation of hepatic arterial infusion of adriamycin. Cancer Res. 1979;39(10):4105-4110.
45. Damodar G, Smitha T, Gopinath S, Vijayakumar S, Rao Y. An evaluation of hepatotoxicity in breast cancer patients receiving injection doxorubicin. Ann Med Health Sci Res. 2015;4(1):74-79.
46. Yamashita T, Wang XW. Cancer stem cells in the development of liver cancer. J Clin Invest. 2013;123(5):1911-1918.
47. Golfieri R, Giampalma E, Renzulli M, et al. Randomised controlled trial of doxorubicin-eluting beads vs conventional chemoembolisation for hepatocellular carcinoma. Br J Cancer. 2014;111(2):255-264.
48. Guan Y-S, He Q, Wang M-Q. Transcatheter arterial chemoembolization: history for more than 30 years. ISRN Gastroenterol. 2012.
49. Xie ZB, Ma L, Wang XB, et al. Transarterial embolization with or without chemotherapy for advanced hepatocellular carcinoma: a systematic review. Tumor Biol. 2014;35(9):8451-8459.
50. Marelli L, Stigliano R, Triantos C, et al. Transarterial therapy for hepatocellular carcinoma: which technique is more effective? A systematic review of cohort and randomized studies. Cardiovasc Intervent Radiol. 2007;30(1):6-25.
51. Hong K, Khwaja A, Liapi E, Torbenson MS, Georgiades CS, Geschwind JF. New intra-arterial drug delivery system for the treatment of liver cancer: preclinical assessment in a rabbit model of liver cancer. Clin Cancer Res. 2006;12(8):2563-2567.
52. Varela M, Real MI, Burrel M, et al. Chemoembolization of hepatocellular carcinoma with drug eluting beads: efficacy and doxorubicin pharmacokinetics. J Hepatol. 2007;46(3):474-481.
53. Huang K, Zhou Q, Wang R, Cheng D, Ma Y. Doxorubicin‐eluting beads versus conventional transarterial chemoembolization for the treatment of hepatocellular carcinoma. J Gastroenterol Hepatol. 2014;29(5):920-925.
54. Gao S, Yang Z, Zheng Z, et al. Doxorubicin-eluting bead versus conventional TACE for unresectable hepatocellular carcinoma: a meta-analysis. Hepatogastroenterology. 2013;60(124):813-820.
55. European Comission. Commission Directive 2009/120/EC of 14 September 2009 amending Directive 2001/83/EC of the European Parliament and of the Council on the Community code relating to medicinal products for human use as regards advanced therapy medicinal products. Off J Eur Union. 2009;52:3-12.
56. Duan F, Lam MG. Delivery approaches of gene therapy in hepatocellular carcinoma. Anticancer Res. 2013;33(11):4711-4718.
57. Mariani JA, Kaye DM. Delivery of gene and cellular therapies for heart disease. J Cardiovasc Transl Res. 2010;3(4):417-426.
58. Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127(7):1323-1334.
59. Wang Z, Sun Y. Targeting p53 for novel anticancer therapy. Transl Oncol. 2010;3(1):1-12.
60. Knapinska AM, Irizarry-Barreto P, Adusumalli S, Androulakis I, Brewer G. Molecular mechanisms regulating mRNA stability: physiological and pathological significance. Curr Genomics. 2005;6(6):471-486.
61. Tuschl T. RNA interference and small interfering RNAs. Chembiochem. 2001;2(4):239-245.
62. Zeng Y, Yi R, Cullen BR. MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proc Natl Acad Sci U S A. 2003;100(17):9779-9784.
63. Walther W, Fichtner I, Schlag PM, Stein US. Nonviral jet-injection technology for intratumoral in vivo gene transfer of naked DNA. In: Walther W, Stein US, eds. Gene Therapy of Cancer: Methods and Protocols, vol 542. New York: Humana Press, 2009:195-208.
64. Hatefi A, Canine BF. Perspectives in vector development for systemic cancer gene therapy. Gene Ther Mol Biol. 2009;13(A):15-19.
65. Clément N, Knop DR, Byrne BJ. Large-scale adeno-associated viral vector production using a herpesvirus-based system enables manufacturing for clinical studies. Hum Gene Ther. 2009;20(8):796-806.
66. Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4(5):346-358.
67. Pack DW, Hoffman AS, Pun S, Stayton PS. Design and development of polymers for gene delivery. Nat Rev Drug Discov. 2005;4(7):581-593.
68. Laquerre S, Anderson DB, Stolz DB, Glorioso JC. Recombinant herpes simplex virus type 1 engineered for targeted binding to erythropoietin receptor-bearing cells. J Virol. 1998;72(12):9683-9697.
69. Trubetskoy VS, Loomis A, Hagstrom JE, Budker VG, Wolff JA. Layer-by-layer deposition of oppositely charged polyelectrolytes on the surface of condensed DNA particles. Nucleic Acids Res. 1999;27(15):3090-3095.
70. Zhu L, Mahato RI. Lipid and polymeric carrier-mediated nucleic acid delivery. Expert Opin Drug Deliv. 2010;7(10):1209-1226.
71. Glover DJ, Glouchkova L, Lipps HJ, Jans DA. Overcoming barriers to achieve safe, sustained and efficient non-viral gene therapy. Adv Gene Mol Cell Ther. 2007;1(2):126-140.
72. Schmidt-Wolf GD, Schmidt-Wolf IG. Non-viral and hybrid vectors in human gene therapy: an update. Trends Mol Med. 2003;9(2):67-72.
73. Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Del Rev. 2011;63(3):131-135.
74. Han S, Mahato RI, Sung YK, Kim SW. Development of biomaterials for gene therapy. Mol Ther. 2000;2(4):302-317.
75. Verma A, Stellacci F. Effect of surface properties on nanoparticle–cell interactions. Small. 2010;6(1):12-21.
76. Guerrero-Cázares H, Tzeng SY, Young NP, Abutaleb AO, Quiñones-Hinojosa A, Green JJ. Biodegradable polymeric nanoparticles show high efficacy and specificity at DNA delivery to human glioblastoma in vitro and in vivo. ACS Nano. 2014;8(5):5141- 5153.
77. Tzeng SY, Higgins LJ, Pomper MG, Green JJ. Biomaterial-mediated cancer-specific DNA delivery to liver cell cultures using synthetic poly (beta-amino esters). J Biomed Mater Res A. 2013;101(7):1837–1845.
78. Poon Z, Lee JB, Morton SW, Hammond PT. Controlling in vivo stability and biodistribution in electrostatically assembled nanoparticles for systemic delivery. Nano Lett. 2011;11(5):2096-2103.
79. Jeong G-J, Byun H-M, Kim JM, et al. Biodistribution and tissue expression kinetics of plasmid DNA complexed with polyethylenimines of different molecular weight and structure. J Control Release. 2007;118(1):118-125.
80. Harris TJ, Green JJ, Fung PW, Langer R, Anderson DG, Bhatia SN. Tissue-specific gene delivery via nanoparticle coating. Biomaterials. 2010;31(5):998-1006.
81. Sunshine JC, Bishop CJ, Green JJ. Advances in polymeric and inorganic vectors for nonviral nucleic acid delivery. Ther Deliv. 2011;2(4):493-521.
82. Wu XZ, Xie GR, Chen D. Hypoxia and hepatocellular carcinoma: The therapeutic target for hepatocellular carcinoma. J Gastroenterol Hepatol. 2007;22(8):1178-1182.
83. Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis-the p53 network. J Cell Sci. 2003;116(20):4077-4085.
84. Hernandez-Alcoceba R, Sangro B, Prieto J. Gene therapy of liver cancer. Ann Hepatol. 2007;6(1):5-14.
85. Hirohashi S. Pathology and molecular mechanisms of multistage human hepatocarcinogenesis. Princess Takamatsu Symp. 1991;22:87-93.
86. Kondo M, Marusawa H, Ueda Y, et al. Diverse p53 gene aberration in hepatocellular carcinoma detected by dual‐color fluorescence in situ hybridization. J Gastroenterol Hepatol. 2004;19(9):1066-1073.
87. Laurent-Puig P, Zucman-Rossi J. Genetics of hepatocellular tumors. Oncogene. 2006;25(27):3778-3786.
88. Guan YS, La Z, Yang L, He Q, Li P. p53 gene in treatment of hepatic carcinoma: status quo. World J Gastroenterol. 2007;13(7):985-992.
89. Anderson SC, Johnson DE, Harris MP, et al. p53 gene therapy in a rat model of hepatocellular carcinoma: intra-arterial delivery of a recombinant adenovirus. Clin Cancer Res. 1998;4(7):1649-1659.
90. Guo Y, Zeng Y, Wang K, et al. [Therapeutic potential of recombinant adenovirus expressing p53 in hepatocellular carcinoma cell lines]. Zhonghua Gan Zang Bing Za Zhi. 2001;9:43-45.
91. Li YX, Lin ZB, Tan HR. Wild type p53 increased chemosensitivity of drug-resistant human hepatocellular carcinoma Bel7402/5-FU cells. Acta Pharmacol Sin. 2004;25(1):76-82.
92. Sangkhathat S, Kusafuka T, Miao J, et al. In vitro RNA interference against β-catenin inhibits the proliferation of pediatric hepatic tumors. Int J Oncol. 2006;28(3):715-722.
93. Zhang R, Ma L, Zheng M, et al. Survivin knockdown by short hairpin RNA abrogates the growth of human hepatocellular carcinoma xenografts in nude mice. Cancer Gene Ther. 2010;17(4):275-288.
94. Li H, Fu X, Chen Y, et al. Use of adenovirus-delivered siRNA to target oncoprotein p28 GANK in hepatocellular carcinoma. Gastroenterology. 2005;128(7):2029-2041.
95. Zhang Y, Ma H, Zhang J, Liu S, Liu Y, Zheng D. AAV-mediated TRAIL gene expression driven by hTERT promoter suppressed human hepatocellular carcinoma growth in mice. Life Sci. 2008;82(23):1154-1161.
96. Gerolami R, Uch R, Faivre J, et al. Herpes simplex virus thymidine kinase-mediated suicide gene therapy for hepatocellular carcinoma using HIV-1-derived lentiviral vectors. J Hepatol. 2004;40(2):291-297.
97. Rajab K, Nelson P, Keung EZ, Conrad C. Suicide Gene Therapy against Cancer. J Genet Syndr Gene Ther. 2013;4(9):1-8.
98. Lemke J, Von Karstedt S, Zinngrebe J, Walczak H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014;21(9):1350-1364.
99. Amano S, Gu C, Koizumi S, Tokuyama T, Namba H. Timing of ganciclovir administration in glioma gene therapy using HSVtk gene-transduced mesenchymal stem cells. Cancer Genomics Proteomics. 2011;8(5):245-250.
100. Bhatia S, Menezes ME, Das SK, et al. Innovative approaches for enhancing cancer gene therapy. Discov Med. 2013;15(84):309-317.
101. Robson T, Hirst DG. Transcriptional targeting in cancer gene therapy. J Biomed Res Int. 2003(2):110-137.
102. Hu BG, Liu LP, Chen GG, et al. Therapeutic efficacy of improved α-fetoprotein promoter-mediated tBid delivered by folate-PEI600-cyclodextrin nanopolymer vector in hepatocellular carcinoma. Exp Cell Res. 2014;324(2):183-191.
103. Hervé J, Cunha AS, Liu B, et al. Internal radiotherapy of liver cancer with rat hepatocarcinoma-intestine-pancreas gene as a liver tumor-specific promoter. Hum Gene Ther. 2008;19(9):915-926.
104. Mohr L, Yoon S-K, Eastman SJ, et al. Cationic liposome-mediated gene delivery to the liver and to hepatocellular carcinomas in mice. Hum Gene Ther. 2001;12(7):799-809.
105. Ahn BC, Ronald JA, Kim YI, et al. Potent, tumor-specific gene expression in an orthotopic hepatoma rat model using a Survivin-targeted, amplifiable adenoviral vector. Gene Ther. 2011;18(6):606-612.
106. Luo SH, Zheng CS, Feng GS, et al. [Experimental studies of rAd-p53 injection by interventional approach for the treatment of rabbit VX2 liver cancer]. Zhonghua Gan Zang Bing Za Zhi. 2010;18(7):502-505.
107. Kim YI, Chung JW, Park JH, Han JK, Hong JW, Chung H. Intraarterial gene delivery in rabbit hepatic tumors: transfection with nonviral vector by using iodized oil emulsion 1. Radiology. 2006;240(3):771-777.
108. Bilbao R, Bustos M, Alzuguren P, et al. A blood-tumor barrier limits gene transfer to experimental liver cancer: the effect of vasoactive compounds. Gene Ther. 2000;7(21):1824-1832.
109. Pearson S, Jia H, Kandachi K. China approves first gene therapy. Nat Biotechnol. 2004;22(1):3-4.
110. Chen S, Chen J, Xi W, Xu W, Yin G. Clinical therapeutic effect and biological monitoring of p53 gene in advanced hepatocellular carcinoma. Am J Clin Oncol. 2014;37(1):24-29.
111. Tian G, Liu J, Zhou JSR, Chen W. Multiple hepatic arterial injections of recombinant adenovirus p53 and 5-fluorouracil after transcatheter arterial chemoembolization for unresectable hepatocellular carcinoma: a pilot phase II trial. Anticancer Drugs. 2009;20(5):389-395.
112. Guan YS, Liu Y, He Q, et al. p53 gene therapy in combination with transcatheter arterial chemoembolization for HCC: one-year follow-up. World J Gastroenterol. 2011;17(16):2143–2149.
113. Dong J, Li W, Dong A, et al. Gene therapy for unresectable hepatocellular carcinoma using recombinant human adenovirus type 5. Med Oncol. 2014;31(8):1-8.
114. Shen A, Liu S, Yu W, Deng H, Li Q. p53 Gene Therapy‐Based Transarterial Chemoembolization for Unresectable Hepatocelluar Carcinoma: A Prospective Cohort Study. J Gastroenterol Hepatol. 2015;30(11):1651-1656.
115. Marshall E. Gene therapy death prompts review of adenovirus vector. Science. 1999;286(5448):2244-2245.
116. Check E. Gene therapy: a tragic setback. Nature. 2002;420(6912):116-118.
117. Deakin CT, Alexander IE, Kerridge I. Accepting risk in clinical research: is the gene therapy field becoming too risk-averse? Mol Ther. 2009;17(11):1842-1848.
118. Sun Yat-sen University. TACE plus recombinant human adenovirus for hepatocellular carcinoma. Clinical trials identification number NCT01869088. https://clinicaltrials.gov/ct2/show/NCT01869088.
119. Shenzhen SiBiono GeneTech Co. Trans-catheter chemo-embolization combined with rad-p53 gene injection in treatment of advanced hepatocellular carcinoma. Clinical trials identification number NCT02418988. https://clinicaltrials.gov/ct2/show/NCT02418988.
