

ADVERTISEMENT
Teenager With Facial Rash, Hypomelanotic Macules, and History of Seizure
What’s Your Diagnosis?
Sharpen Your Physical Diagnostic Skills

HISTORY
A 15-year-old girl presented with yellowish papules on the cheeks and nose. The papules, which first appeared at age 5 years, had become more prominent and profuse in the past 4 years. The patient had recurrent episodes of generalized tonic-clonic seizures from age 2 to 6 years but had been seizure-free since then. She had no headache, chest pain, shortness of breath, or hematuria. Her intelligence was normal. There was no family history of similar skin lesions or seizure disorder.
PHYSICAL EXAMINATION
Vital signs normal. Multiple small, skin-colored papules noted over the nose and both cheeks; 4 hypomelanotic macules and 7 café au lait spots present on the trunk. Remaining physical examination findings unremarkable.
WHAT'S YOUR DIAGNOSIS?
Answer on next page
What's Your Diagnosis?
 ANSWER: TUBEROUS SCLEROSIS COMPLEX
ANSWER: TUBEROUS SCLEROSIS COMPLEX EPIDEMIOLOGY AND ETIOLOGY
TSC is an inherited neurocutaneous multisystem disorder characterized by the development of hamar-tomas in almost every organ, notably in the skin, brain, kidneys, heart, and eyes.1 TSC affects both sexes and all ethnic groups. The prevalence is estimated to be 1 case per 6000 to 10,000 children.1
TSC is caused by a mutation in TSC1 or TSC2; genes that encode for hamartin and tuberin, respectively.1,2 TSC has an autosomal dominant mode of inheritance with almost complete penetrance but variable expressivity.3 Approximately 70% of cases are caused by spontaneous mutations.4,5
 Both TSC1 and TSC2 have tumor suppressor activity, which when inactivated, leads to uncontrolled cell cycle progression and the proliferation of hamartomas throughout the body.1 Mutations in TSC2 are 4 to 5 times more common than those in TSC1 and generally result in a more severe phenotype.2
Both TSC1 and TSC2 have tumor suppressor activity, which when inactivated, leads to uncontrolled cell cycle progression and the proliferation of hamartomas throughout the body.1 Mutations in TSC2 are 4 to 5 times more common than those in TSC1 and generally result in a more severe phenotype.2
CLINICAL MANIFESTATIONS
The Table provides a summary of the primary manifestations of TSC in various organ systems and the frequency of their occurrence.6-19
DIAGNOSIS
Diagnosis of TSC requires the presence of 2 major features or 1 major feature plus 2 minor features.20 Additional diagnostic categories include probable TSC (1 major feature plus 1 minor feature) and possible TSC (1 major feature or 2 or more minor features).20 The presence of hamartomas in 2 different organ systems is considered by some clinicians to be sufficient for diagnosis.
The major features are:
•Facial angiofibromas or forehead plaque.
•Nontraumatic ungual or periungual fibroma.
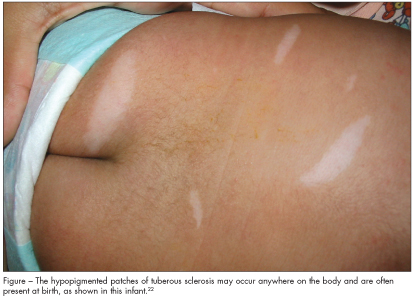
•Hypomelanotic macules—more than 3.
•Shagreen patch (connective tissue nevus).
•Cortical tuber.
•Subependymal nodule.
•Subependymal giant cell astrocytoma.
•Multiple retinal nodular hamartomas.
•Cardiac rhabdomyoma, single or multiple.
•Pulmonary lymphangioleiomyomatosis.
•Renal angiomyolipoma.
In patients with both lymphangioleiomyomatosis and renal angiomyolipomas, other features of TSC should be present before a definite diagnosis is assigned.20
The minor features are:
•Multiple randomly distributed pits in dental enamel.
•Hamartomatous rectal polyps (histologic confirmation is suggested).
•Bone cysts (radiologic confirmation is suggested).
•Cerebral white matter migration tracts (radiologic confirmation is suggested).
•Gingival fibromas.
•Nonrenal hamartoma (histologic confirmation is suggested).
•Retinal achromic patch.
•“Confetti” skin lesions.
•Multiple renal cysts (histologic confirmation is suggested).
When a cortical tuber and cerebral white matter migration tracts occur together, they should be counted as 1 rather than 2 features of TSC.20
 DIAGNOSTIC STUDIES
DIAGNOSTIC STUDIES
The following tests are recommended in the initial evaluation and in follow-up care of patients with TSC:
•Echocardiography to check for cardiac rhabdomyomas and other cardiac anomalies.
•ECG to identify arrhythmias.
•MRI or CT scans of the brain for confirmatory evidence of TSC, such as cortical tubers.
•Renal ultrasonography is performed at the time of diagnosis and repeated every 1 to 3 years, depending on the level of concern.7,19 Monitoring of renal function is important in those children with structural renal abnormalities. Small angiomyolipomas usually do not cause symptoms; lesions larger than 4 cm are associated with an increased risk of serious hemorrhage, which may present as flank pain or hematuria.8 Renal tumors may also obstruct urine outflow or distort renal parenchyma.21
•Chest and skeletal radiographs can be ordered when indicated.
•Molecular genetic studies for TSC1 and TSC2 mutations are helpful in patients who do not meet the criteria for definite TSC and in clinically normal family members of an affected child to improve genetic counseling.
MANAGEMENT
Treatment is symptomatic and organ-specific and focuses on improving patient outcome and quality of life. Patients with TSC benefit from a multidisciplinary approach. Early intervention is important for the family—to help parents organize the most appropriate educational placement, when necessary, and plan specialist referral and follow-up.
Facial angiofibromas and forehead plaques, the
potentially embarrassing cosmetic stigmata of TSC, can be treated with dermabrasion, laser surgery, or cryosurgery.9 Shagreen patches can be treated with laser surgery, shave excision, or dermabrasion.
Of the available antiepileptic medications, vigabatrin is often used as first-line therapy and has been shown to be especially effective for infantile spasms and partial seizures.10 Consider neurosurgical intervention for patients with intractable seizures, hydrocephalus, increased intracranial pressure, and interval or rapid enlargement of a subependymal giant cell astrocytoma or neurological deficits attributable to the tumor.1
References
1.Franz DN, Bissler JJ, McCormack FX. Tuberous sclerosis complex: neurological, renal and pulmonary manifestations. Neuropediatrics. 2010;41:199-208.
2. Orlova KA, Crino PB. The tuberous sclerosis complex. Ann N Y Acad Sci. 2010;1184:87-105.
3. Leung AK, Robson WL. Tuberous sclerosis complex: a review. J Pediatr Health Care.2007;21:108-114.
4. de Vries PJ. Targeted treatments for cognitive and neurodevelopmental disorders in tuberous sclerosis complex. Neurotherapeutics. 2010;7:275-282.
5. Camposano SE, Greenberg E, Kwiatkowski DJ, Thiele EA. Distinct clinical characteristics of tuberous sclerosis complex patients with no mutation identified. Ann Hum Genet. 2009;73:141-146.
6. Sweeney SM. Pediatric dermatologic surgery: a surgical approach to the cutaneous features of tuberous sclerosis complex. Adv Dermatol. 2004;20:117-135.
7. Leung AK, Kamat D. Tuberous sclerosis complex. In: Kimura R, ed. Genetic Inheritance Patterns.New York: Nova Biomedical Books; 2008:271-289.
8. Roach ES, Sparagana SP. Diagnosis of tuberous sclerosis complex. J Child Neurol. 2004;19:643-649.
9. Schwartz RA, Fernández G, Kotulska K, Józ´wiak S. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. J Am Acad Dermatol. 2007;57:189-202.
10. Moavero R, Cerminara C, Curatolo P. Epilepsy secondary to tuberous sclerosis: lessons learned and current challenges. Childs Nerv Syst. 2010;26:1495-1504.
11. Yates JR. Tuberous sclerosis. Eur J Hum Genet. 2006;14:1065-1073.
12. Kandt RS. Tuberous sclerosis complex and neurofibromatosis type 1: the two most common neurocutaneous diseases. Neurol Clin. 2003;20:983-1004.
13. Curatolo P, Napolioni V, Moavero R. Autism spectrum disorders in tuberous sclerosis: pathogenetic pathways and implications for treatment. J Child Neurol. 2010;25:873-880.
14. Numis AL, Major P, Montenegro MA, et al. Identification of risk factors for autism spectrum disorders in tuberous sclerosis complex. Neurology. 2011;76:981-987.
15. D’Agati E, Moavero R, Cerminara C, Curatolo P. Attention-deficit hyperactivity disorder (ADHD) and tuberous sclerosis complex. J Child Neurol. 2009;24:1282-1287.
16. Hottinger AF, Khakoo Y. Neurooncology of familial cancer syndromes. J Child Neurol.2009;24:1526-1535.
17. Ertan G, Arulrajah S, Tekes A, et al. Cerebellar abnormality in children and young adults with tuberous sclerosis complex: MR and diffusion weighted imaging findings. J Neuroradiol.2010;37:231-238.
18. Chu-Shore CJ, Major P, Montenegro M, Thiele E. Cyst-like tubers are associated with TSC2 and epilepsy in tuberous sclerosis complex. Neurology. 2009;72:1165-1169.
19. Borkowska J, Schwartz RA, Kotulska K, Jozwiak S. Tuberous sclerosis complex: tumors and tumorigenesis. Int J Dermatol. 2011;50:13-20.
20. Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol. 1998;13:624-628.
21. Rakowski SK, Winterkorn EB, Paul E, et al. Renal manifestations of tuberous sclerosis complex: incidence, prognosis, and predictive factors. Kidney Int.& 2006;70:1777-1782.
22. Defendi GL. Hypopigmented patches in infant with a history of seizures. Consultant For Pediatricians. 2008;7:513-520.










