ADVERTISEMENT
Managed Care Implications for Biosimilars
Tampa—While the demand for biologic agents continues to increase, biosimilars are gaining momentum as follow-on reproductions of biologics. The first FDA-approved biosimilar is expected to reach the US market in 2015, though they have been on the European market since 2006. A satellite symposium at the AMCP meeting examined the approval process and managed care implications for biosimilars reaching the market. This satellite symposium was supported by an educational grant from Hospira, Inc.
Ali McBride, PharmD, MS, BCPS, clinical coordinator of hematology/oncology, University of Arizona Cancer Center, Tucson, Arizona, began the session with an introduction on biosimilars. These are biologic products that are demonstrated to be “highly similar” to an FDA-approved biologic product. New licensure pathways permit a biosimilar biologic product to be licensed based on less-than-full complement of product-specific, nonclinical, and clinical data.
An Understanding of Biotechnology
Biotechnology growth and development provides new approaches to discovery, design, and production of new drugs, according to Dr. McBride. Biotechnology makes the following possible: prevention, cure, and treatment of more diseases; targeted, more effective, less toxic medications; proactive versus reactive approach;p roduction of replacement human proteins; and production of “pure” drugs (no contamination by infectious pathogens from human or animal sources).
The FDA stipulates that pharmaceutically equivalent drug products be formulated to contain the same amount of active ingredient in the same dose form and meet the same or compendial or other applicable standards (ie, strength, quality, purity, and identity). Products are considered therapeutic equivalents if they are pharmaceutical equivalents and can be expected to have the same clinical effect and safety profile when administered to patients under the conditions specified in the labeling. However, new biotechnology drugs may not be therapeutically equivalent to the originator and may infringe on 1 or more of the originator’s patents.
Biosimilar Approvals
Carina Dolan, PharmD, BCOP, senior clinical manager for oncology, clinical solutions team, Novation, Irving, Texas, continued the presentation by discussing the clinical and formulary implications of biosimilars coming to the US market. Healthcare stakeholders will need to understand approval pathways, drug interchangeability, and cost-saving strategies, presenting both opportunities and challenges for managed care professionals.
FDA guidance documents on biosimilars were first published on February 9, 2012, including quality and scientific considerations in demonstrating biosimilarity of drugs. Five new guidance documents are planned for release in 2014, which will also include clinical pharmacology data to support demonstration of biosimilarity, considerations in demonstrating interchangeability with a reference product, labeling for biosimilar biologic products, and reference product exclusivity for 351(a)-filed biologic products.
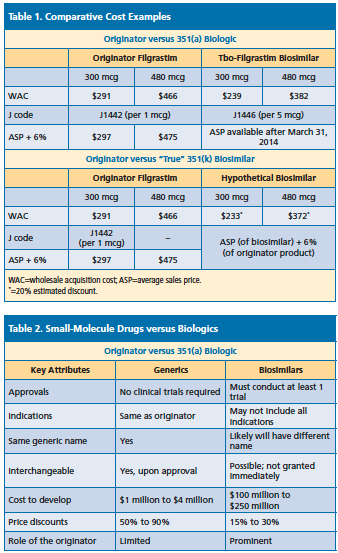
Once the FDA accepts a biosimilar agent, that information must be shared with the originator company within 20 days. Dr. Dolan said the key question would be whether biosimilar applicants use the 351(k) pathway or file a full Biologics License Application (See Table 1 below).
Currently, there is controversy over whether a biosimilar agent should have a unique name or if it should have the same name as its originator. Companies in support of the unique name include the Biotechnology Industry Organization, Amgen, Janssen Pharmaceuticals, and Pfizer. Companies in support of using the same name as the originator include the Generic Pharmaceutical Association, Hospira, Novartis, and Mylan.
Dr. Dolan noted that the adoption of biosimilars will be more complex than generic medications (See Table 2 below). Unlike generics, pharmacy and therapeutics committee involvement is necessary, there must be a detailed evaluation of safety and efficacy, and mechanics for prescribing, administration, and documentation are required.
Managing Biosimilars
Dr. Dolan concluded by listing key questions that will arise for formulary management with regard to biosimilar approvals and implementations:
• What was the approval history of the biosimilar?
• What information is available concerning the clinical efficacy and safety of the biosimilar (eg, FDA review document, published trials, European data, AMCP dossier, expert organization guidelines)?
• Will the biosimilar product be endorsed only for labeled indications or for off-label indications as well?
• What is the existing level of adverse events with the originator product?
• What modifications need to be made to existing order sets and protocols to
include biosimilar products?
• What education will need to be provided to physicians, nurses, and other
clinicians to prepare for biosimilar adoption?
• What patient education materials will be needed to support biosimilar use?
• What is the financial value associated with use of a biosimilar (comparative
cost and reimbursement)?
Overall, biosimilar adoption will be more complex and will require education among pharmacists, physicians, patients, and other players in the healthcare arena. Healthcare organizations will need to invest more resources to evaluate and incorporate biosimilars into practice.