

Concerns of the Emerging Biosimilars Market
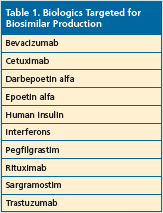
The anticipated arrival of the first biosimilar in the United States came on March 6, 2015, with the FDA’s approval of ZarxioTM (filgras- tim-sndz). Manufactured by Sandoz, a Novartis company, Zarxio is a copy of Amgen’s Neupogen® (filgrastim)—a white blood cell booster.1 This is the first in what is expected to be an expanding market of biosimilars as many patents on biologics are scheduled to expire soon. Over the next several years, many biologic products will be targeted to serve as substitute biosimilars (Table 1).2
 Although the US healthcare industry has been grappling with a number of issues regarding the adoption of biosimilars prior to the first FDA approval, now biosimilars are a reality and the need for clarity on the issues surrounding the market is even more pressing.
Although the US healthcare industry has been grappling with a number of issues regarding the adoption of biosimilars prior to the first FDA approval, now biosimilars are a reality and the need for clarity on the issues surrounding the market is even more pressing.
The entire healthcare industry, particularly managed care, stands to reap a significant benefit from adopting biosimilars given their expected re- duction in cost. According to a 2014 analysis by the RAND Corporation, biosimilars are predicted to reduce US spending on biologics by $44.2 billion over the next decade.3
Among the issues of primary importance for managed care in adopting biosimilars into clinical practice is the need to ensure that these products are both safe and effective. In addition, how to label these products is also an area of concern and one that promises to influence the adoption of biosimilars.
Efficacy and Safety
A biosimilar is a copy of an approved original biologic drug, a drug that is produced and isolated from living systems (eg, yeast, bacteria, mammalian cells).4,5 Similar to the reason for the development of generic drugs, which is to offer a lower cost version of an original drug whose patent protection has expired, biosimilars offer a similar biologic drug to its originator biologic once the data protection on the originator has expired.4,5 Unlike generic drugs, however, biosimilars are not identical copies of the original biologic drug given the inherent variability of the biologic system and manufacturing process.4
Because biosimilars are not entirely identical to their originator product, a major concern is how to determine their efficacy and saftey. Guidance comes from the FDA, which has stipulated that a biosimilar can only be approved if it is highly similar to the FDA-approved biological product (referred to as the reference product) with no clinically meaningful differences in safety and effectiveness from the reference product.6
The FDA also can approve a biosimilar application as an “interchangeable bio- logic” that basically meets the requirements for approval for a biosimilar (highly similar to the reference product without any meaningful clinical differences) but with the additional evidence showing comparable or better safety and efficacy when the “interchangeable biologic” is substituted for the reference biologic.7 In addition, under the Biologics Price Competition and Innovation Act of 2009, which is part of the Affordable Care Act, an “interchangeable biologic” can be substituted for the reference product without the healthcare provider who prescribed the reference product intervening.1 The acceptance of biosimilars as substitutions for the reference biologic, however, is subject to state laws. To date, 23 states have considered legislation on establishing state standards for biosimilar substitution. Table 2 lists several common aspects to this legislation.8
 In the approval of Zarxio, for example, the FDA relied on a broad range of safety and effectiveness evidence (ie, laboratory and animal study data, along with human pharmacokinetic, pharmacodynamic, and clinical immunogenicity data) that it felt demonstrated that Zarxio was biosimilar to Neupogen. However, the FDA did not approve it as an “interchangeable biologic.”
In the approval of Zarxio, for example, the FDA relied on a broad range of safety and effectiveness evidence (ie, laboratory and animal study data, along with human pharmacokinetic, pharmacodynamic, and clinical immunogenicity data) that it felt demonstrated that Zarxio was biosimilar to Neupogen. However, the FDA did not approve it as an “interchangeable biologic.”
An important component of establishing the safety of the biosimilars will be an ongoing assessment with the FDA planning to monitor post approval safety based on the FDA Adverse Event Reporting System.
It has also been suggested that managed care and other healthcare organizations should conduct their own post-marketing drug safety using real-world, independent data to ensure that potential adverse effects are tracked.9
Product Name
An issue under debate that may affect the adoption of biosimilars by managed care and other healthcare professionals is how biosimilars are labeled—should they be given the same nonproprietary name as the originator biologic or a dif- ferent name? A survey published in the Journal of Managed Care & Specialty found that about 75% of pharmacists surveyed would feel confident substituting an interchangeable biosimilar to its branded biologic if both the biosimilar and biologic shared the same nonproprietary name, whereas only 25.3% of pharmacists would feel confident if the products had a different name.10 As of yet, the FDA has not provided guidance on how biological products should be named.1
Summary
As biosimilars come to the US market, several challenges will be faced by managed care and other healthcare professionals in regards to adopting these lower-cost biologic alternatives in clinical practice. Regulations by the FDA and post-surveillance safety data based on real-world experience will help provide the necessary guidance on the safety and effectiveness of these drugs on an ongoing basis.
References
1. U.S. Food and Drug Administration. FDA approves first biosimilar product Zarxio. March 6, 2015. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm436648.htm. Accessed June 2, 2015.
2. Fitzgerald K. Biosimilars coming to market in lenges and risks. Swiss Med Wkly. 2014;144. 2015, questions ensue. First Report Managed Care. 2014;8466. www.firstreportnow.com/articles/biosimilars-coming-market-2015-questions-ensue. Accessed June 2, 2015.
3. RAND Corporation. Biosimilar medications could create billions in health care savings. Press Release. November 3, 2014. www.rand.org/news/press/2014/11/03.html. Accessed June 2, 2015.
4. Weise M, Bielsky MC, De Smet K, et al. Biosimilars: what clinicians should know. Blood. 2012;120(26):5111-5117.
5. Müller R, Renner C, Gabay C. Cassata G, Lohri A, Hasler P. The advent of biosimilars: challenges and risks. Swiss Med Wkly. 2014;144.
6. U.S. Food and Drug Administration. Information for Healthcare Professionals (Biosimilars). https://1.usa.gov/1cvucwO. Accessed June 2, 201.5
7. Reinke T. Biosimilars charge the market but still face many hurdles. First Report Managed Care. April 2015. www.managedcaremag.com/archives/2015/4/biosimilars-charge-market-still-face-many-hurdles. Accessed June 2, 2015.
8. Cauchi R. State laws and legislation related to biologic medications and substitution of biosimilars. National Conference of State Legislation. April 29, 2015. https://www.ncsl.org/research/health/state-laws-and-legislation-related-to-biologic-medications-and-substitution-of-biosimilars.aspx. Accessed June 2, 2015.
9. Davis J. Monitoring the safety of biosimilars by managed care organizations: a practical approach. Adverse Events: Redefining Drug Safety. March 20, 2015. https://rxview.adverseevents.com/monitoring-the-safety-of-biosimilars-by-managed-care-organizations-a-practical-approach. Accessed June 2, 2015.
10. Fernandez-Lopez S, Kazzaz D, Bashir M, McLaughlin T. Assessment of pharmacists’ views on biosimilar naming conventions. J Manag Care Spec Pharm. 2015;21(3):188- 195.
 Gary M. Cohen, BSPharm, RPh, CSP, publisher, Specialty Pharma Journal, spoke with First Report Managed Care to provide some perspective on biosimilars and their potential and probable effect on the managed care field as well as the healthcare industry as a whole. Mr. Cohen has extensive experience in the specialty pharmacy arena, and co-hosted the Biosimilars 20/20 meeting, along with the Specialty Pharma Education Center (SPEC), which was held June 3 to 4 in Philadelphia. The meeting brought together all the major stakeholders of the emerging market of biosimilars.
Gary M. Cohen, BSPharm, RPh, CSP, publisher, Specialty Pharma Journal, spoke with First Report Managed Care to provide some perspective on biosimilars and their potential and probable effect on the managed care field as well as the healthcare industry as a whole. Mr. Cohen has extensive experience in the specialty pharmacy arena, and co-hosted the Biosimilars 20/20 meeting, along with the Specialty Pharma Education Center (SPEC), which was held June 3 to 4 in Philadelphia. The meeting brought together all the major stakeholders of the emerging market of biosimilars.
Q: A major challenge for providers to adopt biosimilars is to determine the efficacy and safety of these agents. How will managed care assess the efficacy and safety of biosimilars?
A: Traditionally, providers have relied on clinical trials to assess efficacy and safety. However, changes in the US market have led to the increasing collection and use of “real-world” evidence, which provides retrospective data on how a product works. Technology is now closing the big gap that has existed between clinical evidence and “real-world” evidence, and this has allowed pharmaceutical manufacturers, payers, and providers access and the ability to look at the real-world evidence. I think biosimilars are going to fall into this pattern and even promote that pattern because there is no clear-cut way to assess biosimilars using the traditional clinical efficacy and safety data. Instead, biosimilars are going to be more apt to rely on real-world evidence of how these products work in terms of not only efficacy and safety, but also in terms of tolerability, cost, and quality of life. So there will be lots of measurements to collect on biosimilars.
The good news for biosimilars is that facilities are already in place for physicians and patients to obtain reported data, as the data collection process has become very important in the whole pharmaceutical industry ecosystem because payers and manufacturers rely on that data.
Managed care needs to know how to collect data and how to use that data, and then how to communicate the biosimilar message downstream to their providers and members so that they can make this something that can become easily adaptable.
The best way to get data is to develop programs that engage patients and providers. If you engage both parties in a program to collect data, that is going to be the key to understanding the real value of these products.
Q: What does managed care need to know about biosimilars in order to adopt them into coverage structures?
A: Physicians need to become well-educated on what biosimilars mean, the differences among them, and the legal and regulatory environment around them. They have to understand that a biosimilar is as good, and potentially even better, than the reference product. Real-world data is available to show this; however, not until a provider sees the comparable or even better efficacy and/or safety of a biosimilar in their actual patients will they know this.
They also need to become educated on the economics of biosimilars. Managed care needs to know what the economic effect will be to their organization and that will drive adoption.
Physicians are going to become educated on biosimilars through big pharma, through tools including predictive analytic models that stratify risk to predict events such as readmission rates and nonadherence rates of patients. Organizations will be doing comparative modeling on these agents, reporting pharmaco- vigilance and adverse events to the FDA or a third party, and will be providing data back to providers, pharma, and payers. Similar to biologics, one biosimilar may work well for one person but not for another and vice versa because of the different chemical makeup of each individual and how they respond to these particular medicines.
Along with education, physicians will also need to gain a comfort level with these drugs and they gain this just as they do with any drug—by prescribing to their patients and see- ing how the patients do on the drug.
Q: You said that providers will need to understand the regulatory and legal environment surrounding biosimilars. Can you expand on this?
A: Biosimilars are still caught between the regulatory environment and legal environment and these will be ongoing challenges to adoption. Biosimi- lars are still in a formative stage and there is no correct blueprint yet for anyone to follow and that makes it a challenge but also creates great opportunities for organizations.
The end game for all stakeholders is first that biosimilars provide a safe, efficacious product that is as good as or better than the reference product in terms of quality of life and clinical response. If that is not in place, than price does not matter. Second, once biosimilars are shown to be as good or better than the reference product, the next step is to establish a cost savings. If we can create a cost savings to the health- care system and reduce cost in the pharmacy benefit market in terms of direct cost for the medication, as well as provide wrap-around services for educating patients and physicians, we can also show additional cost savings on the medical side and then we have victory.
Q: Who or what will drive demand for biosimilars?
A: I do not think consumers will drive demand; contracting will. Physicians are basically told what to prescribe–they follow the formularies that are mandated by their payer contracts. But there is one place that it would behoove physicians to adopt biosimilars, and that is in integrated networks and account- able care organizations because, in these organizations, physicians have skin in the game by the pay-for-performance model that is emerging. These physicians will be more apt to pick a lower cost drug and want to adopt one for cost savings.—Mary Beth Nierengarten



